
单原子催化剂在类芬顿水处理领域的研究进展
2024-03-18 05:54孙天礼黄炳坤熊兆锟
应用化学 2024年2期
孙天礼 朱 国 何 海 黄炳坤 熊兆锟* 赖 波*
1(中国石化西南油气分公司采气二厂,阆中 637400)
2(四川大学建筑与环境学院,中德水环境与健康研究中心,成都 610065)
利用高级氧化工艺(AOPs)处理环境中的难降解污染物是环境修复有效的手段[1-2]。然而,催化剂的活性、稳定性和选择性是影响其实际应用最大的障碍。在过去10年中,具有极高原子分散性的单原子催化剂(SACs)受到广泛关注。而且关于SACs 应用的研究论文数量也成倍增长[3-4]。2011 年,Zhang等[5]将分散的单个Pt原子锚定在FeOx纳米晶体表面,首次提出了SACs,此后大量的研究人员被SACs独特的几何结构和电子特性所吸引。与传统的纳米颗粒催化剂相比,SACs 具有均匀的活性位点,使得对反应机理的研究更便利、更准确,表现出优异的催化活性和稳定性,在能源[6-8]、环境[9-11]、有机合成[12-14]和生物医学[15-17]等领域广泛应用。
随着催化剂尺寸的缩小,比表面积和催化位点密度增大,催化活性得到显著提高[18-21]。过渡金属由于其存在空轨道或未配对电子,更有可能接受氧化剂或底物中的配对电子,通常被认为是催化活性中心[22]。一些综述归纳并总结了SACs 在AOPs 中的应用,提出了SACs 在类芬顿技术中的未来机遇和挑战[11,23]。Kim 等[10]总结了SACs 在过硫酸盐基的AOPs 中的应用,讨论了它们与相应的金属离子和纳米颗粒的功能差异。Xu等[23]揭示了不同AOPs体系中SACs通用催化氧化途径的来源以及碳基SACs上微污染物的降解机制。目前,SACs在高级氧化水处理中的研究正处于快速发展阶段,系统地总结SACs的合成及其在类芬顿中的应用有重要意义。
本文总结了SACs的合成策略及其在AOPs中应用的研究现状:首先,对近10年来SACs的合成方法进行了总结以指导未来SACs的制备;随后,综述了SACs在类芬顿反应中的应用和催化表现,特别是对类芬顿反应中SACs的实际应用进行了总结;最后,针对SACs在AOPs中应用所面临的挑战和未来的研究趋势进行了讨论和展望。
1 SACs合成及表征
1.1 SACs合成方法
制备SACs 需要使金属原子分散于载体上。然而,孤立的金属原子比金属纳米颗粒和团簇具有更高的表面能,在合成过程中容易聚集,给合成SACs 带来了巨大的挑战。尽管大多数研究中提到高温会引起金属颗粒的快速聚集,但Li 等[24]发现在热解过程中有烧结和雾化反应过程,在不同的温度下主导的反应是不同的。如图1a 所示,铂金纳米颗粒首先被包裹在ZIF-8 中。在较低的温度(300~900 ℃)下,烧结占主导地位,铂金纳米颗粒聚集成较大的纳米颗粒。在更高的温度(900~1000 ℃)下,雾化过程占据了主动,较大的铂金纳米颗粒开始分散。最后,形成了具有完全分散的铂原子的Pt-SAC,实现了从纳米颗粒催化剂到单原子催化剂的转变。许多SACs 的制备采用低的金属前驱体添加量来减少金属原子的聚集,但这种方法合成的SACs会存在金属负载量偏低的问题。如何制备高密度SACs仍是一个重大挑战。近年来,越来越多的技术(如原子层沉积、热解和湿化学法)可用于合成稳定的高负载SACs[25-26]。除此之外,随着对催化机理研究的深入,如何精确调控单原子催化剂的配位结构成为新的研究热点。例如,原子层沉积法(ALD)能实现从单原子到纳米颗粒的转变,通过控制ALD 循环次数,可以精确控制金属物种的尺寸、密度和负载。Sun 等[27]通过控制ALD 循环的次数可以很容易地控制Pt物种的大小(图1b)。在50、100和150次循环后,Pt粒子的平均粒径为0.5、1~2和2~4 nm。
1.2 SACs表征方法
识别单个金属原子和确认配位环境对于了解AOPs 过程中的催化机理至关重要。高角环形暗场像-扫描透射电子显微镜(HAADF-STEM)、X 射线吸收精细结构光谱(EXAFS)和穆斯保尔谱已被用于提供单原子存在的直接证据,此外密度泛函理论(DFT)计算常常被用于揭示SACs 的活性位点和电子特征。
HAADF-STEM 的图像强度与原子序数的平方成正比,这使得重金属原子与轻金属原子相比显示出明亮的对比度[28]。Li等[29]通过热解配位聚合物策略合成了含铁量高(质量分数30%)的Fe-SAC。图2a中的亮白点是分散的铁原子,其原子序数较大,对比度较高。通过HAADF-STEM 观察到,虽然铁的负载量特别高,但催化剂中的铁是均匀分散的(用红圈标出),没有观察到纳米铁簇或纳米颗粒。EXAFS可以观察吸收元素的数量和类型,EXAFS 为观察原子提供了一种直接的方法。一般来说,单个金属原子与非金属元素(N、O和S等)之间的键长小于金属与金属之间的键长。因此,可以通过EXAFS中的峰位差异来判断SACs 是否合成成功。Huang 等[30]通过对比FeSA-MNC、Fe 箔、Fe2O3和铁酞菁(FePc)的EXAFS发现FeSA-MNC只在1.52 Å处有一个明显的峰,对应于FePc的Fe-N配位,表明了Fe物种为单分散(图2b)。铁是类芬顿反应重要的活性组分,穆斯保尔谱是表征铁基催化剂的重要方法之一,因此穆斯保尔谱也被常用于表征铁基单原子催化剂,特别是原位动态表征活性位点反应过程中结构和自旋态等的变化。Li等[31]首次建立了可操作的57Fe 穆斯保尔谱光谱,以确定氧还原反应过程中分散的活性铁原子的确切结构和自旋状态。
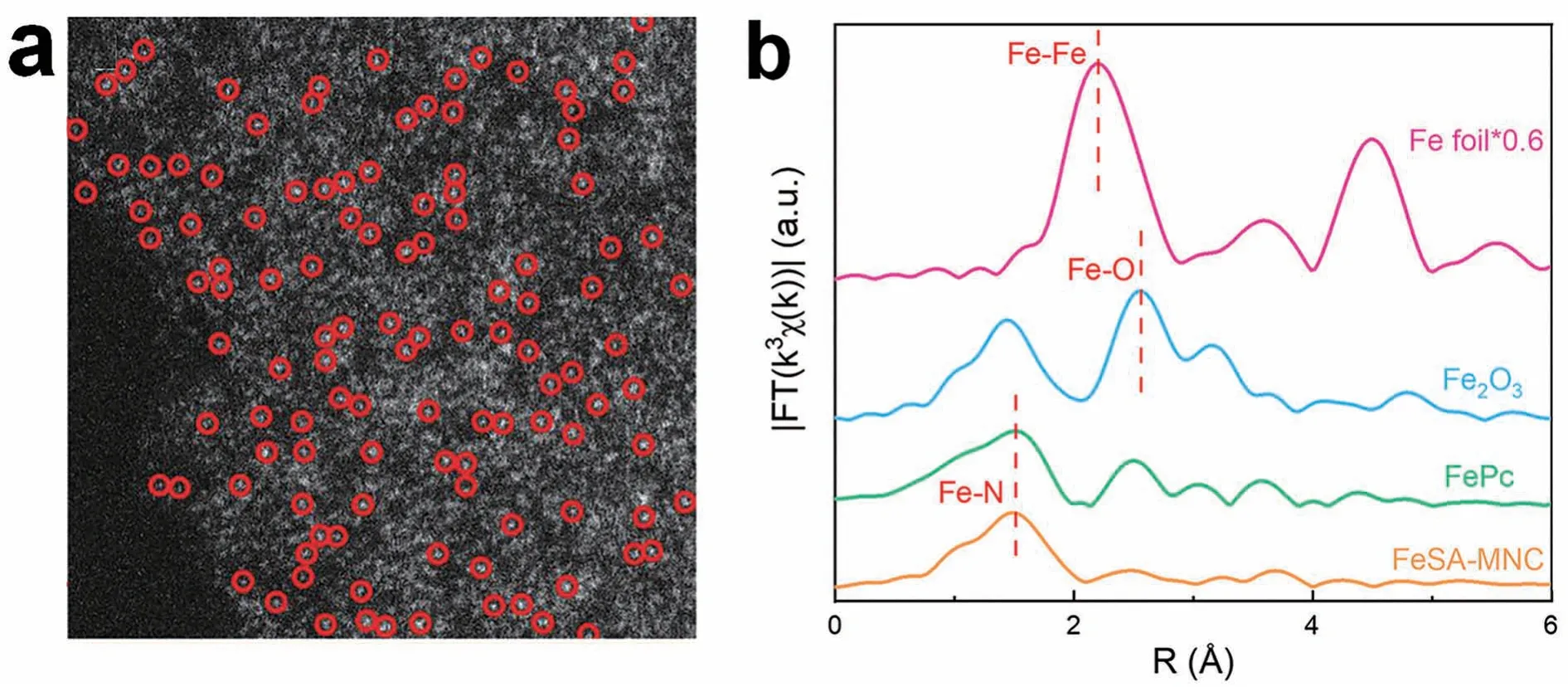
图2 (a)通过SAS-Fe 观测到的HAADF-STEM。比例尺:2 nm[29];(b) FeSA-MNC、Fe 箔、Fe2O3 和FePc 的EXAFS[30]Fig. 2 (a) HAADF-STEM obtained from SAS-Fe.Scale bar:2 nm[29];(b) EXAFS of FeSA-MNC,Fe foil,Fe2O3 and FePc[30]
2 SACs在类芬顿反应中的催化表现
2.1 过一硫酸盐(PMS)
基于PMS 的AOPs 体系可以高效地产生高活性的自由基(•OH 和),多年来一直被用于环境中有机物的去除[32-34]。近年来,越来越多的研究关注SACs 活化PMS 这一领域。Li 等[35]首次将SACs 引入活化PMS 的类芬顿反应中,通过将单个钴原子锚定在氮掺杂的多孔石墨烯上,实现了4 min 内对20 mg/L 双酚A(BPA)的高效降解。Wu 等[36]利用氧/氮共掺杂策略调控钴原子配位结构,识别并制备出具有高效活化过硫酸盐的优势配位构型CoN5O1,研发了以高价钴氧物种为主要活性物种和多种自由基共同作用的新型催化氧化体系CoN5O1/PMS,显著降低了PMS 活化的反应能垒(图3a)。Mo 等[37]利用原子团簇调控了邻近单原子位点的电子配置,构建了缺电子中心,优化了d带中心位置,赋予了催化剂优异的PMS 吸附能和PMS 产物脱附能,同时,团簇位点倾向于起到助催化剂的作用。PMS 在催化剂表面发生氧化反应,还原缺电子Fe 中心,可见光有效加速此反应循环(图3b)。Song 等[38]通过构建局部电场诱导耦合电子-质子转移过程促进高价金属转化的新策略构建了非对称配位的CoB1N3单原子催化剂,该催化剂可形成强极性电场,通过耦合电子-质子转移过程加速了O—H 键的裂解,诱导了O—O 键的断裂和Co—O 间的强电子转移,实现了Co(Ⅳ)O 的高选择性转化(图3c)。
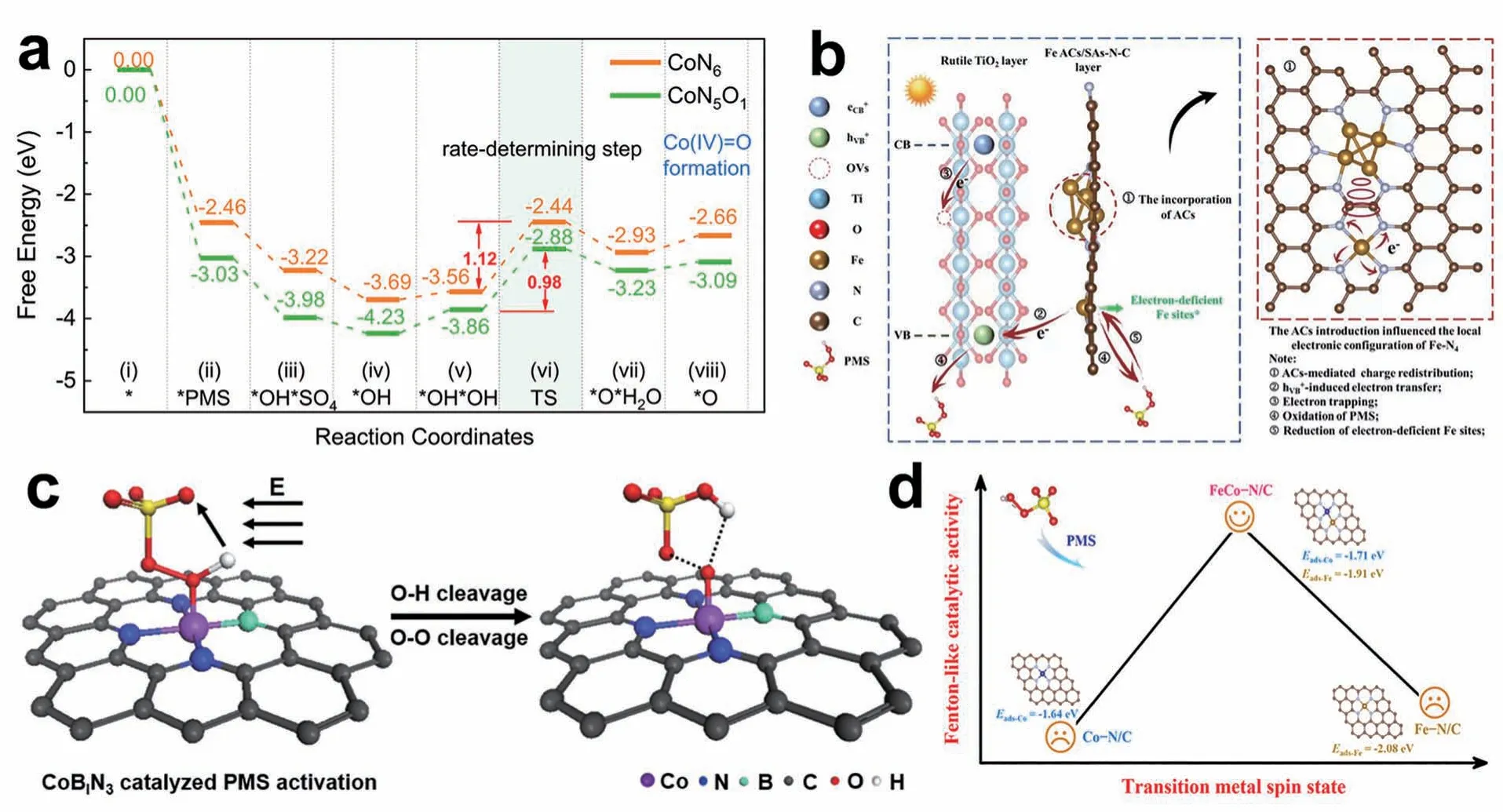
图3 (a) CoN6/PMS 和CoN5O1/PMS 体系Co(Ⅳ)O 生成的能量谱,*O 表示Co(Ⅳ)O 的反应种类[36];(b)提议的Vis/TiFeAS/PMS 体系的反应机制[37];(c)局部电场诱导耦合电子-质子转移过程促进高价金属转化示意图[38];(d)过渡金属自旋态与类芬顿催化活性的关系[39]Fig. 3 (a) Energy profiles of Co(Ⅳ)O formation for CoN6/PMS and CoN5O1/PMS systems;*O represents the reactive species of Co(Ⅳ)O[36];(b) The proposed reaction mechanisms of Vis/TiFeAS/PMS system[37];(c) Schematic illustration of a local electric field-induced coupled electron-proton transfer process promoting the conversion of high-valent metal-oxo species[38];(d) Relationship between spin states and Fenton-like catalytic activity of transition metals[39]
除了含有单一金属的SACs 外,还有含有不同金属的SACs。双原子催化剂在电催化、化学合成和环境修复等催化反应中具有广阔的应用前景。通过将单个钴原子锚定在氮掺杂的多孔石墨烯上,Li等[35]实现了高效的类芬顿催化降解BPA。20 mg/L 的BPA 可在4 min 内被完全降解。自由基淬灭和电子顺磁共振(EPR)实验证明,1O2是主要的活性物质。Co-N4位点有效地促进电荷转移以活化PMS,而BPA 则吸附在相邻的吡咯N 位点上。双反应位点显著减小了PMS 活化产生的1O2对污染物的迁移距离。因此,类芬顿催化活性显著提高。Zhao 等[39]系统地比较了双原子FeCo-N/C 与其单原子对应物通过活化PMS 用于污染物去除的催化性能,来探究双原子催化剂高活性的起源和内在活性增强的机制。FeCo-N/C 的特殊自旋态重构有效地改善了Fe 和Co 在d轨道上的电子结构,提高了PMS 的活化效率(图3d)。结合理论计算发现,与单独Co 原子或Fe 原子向PMS 分子传递电子不同,FeCo-N/C 中的Fe原子向邻近的Co 原子提供额外的电子,并使Co 中心的d带正移,从而优化PMS 的吸附和分解,使其通过低能垒途径生成独特的高价FeⅣ-O-CoⅣ物种。
相对于其它氧化剂,PMS 在类芬顿反应中通常表现出较好的催化性能。SACs 在PMS 的活化中发挥了很好的作用,特别是当掺杂双金属活性位点并与其它先进材料结合时,往往能达到协同催化的效果。但是相对于其它氧化剂体系,PMS基AOPs具有更高催化效率的本质仍有待进一步研究。此外,尽管PMS 的催化效果非常显著,但与其它氧化剂相比,PMS 的价格相对较高,并且存在不能有效地从废水中去除的问题,这限制了PMS基AOPs的实际应用。
2.2 过二硫酸盐(PDS)
与PMS相比,PDS的价格和毒性相对较低[1],且PDS和亚硫酸盐的加入不显著影响水体的pH值[40]。Zuo等[4]设计了一种新型的C3N4-rGO 夹层结构来稳定分散的Fe原子(图4a)。在pH 值(0~14)范围内,C3N4-Fe-rGO/PDS 体系能够在阳光照射下15 min 内完全降解四环素。与C3N4-Fe/PDS 体系相比,加入还原氧化石墨烯后,C3N4-Fe-rGO 催化剂对pH 值的适应性提高,在不同pH 值条件下表现出较高的稳定性和可回收性(图4b)。Wang 等[41]的研究表明相邻Cu 原子的距离(dCu1-Cu1)与反应物PDS 分子大小的匹配程度决定了碳负载SACs 上的类芬顿反应的活性大小(图4c)。当dCu1-Cu1在5~6 Å 范围内,与PDS的分子大小相匹配,其转化率比其它范围的dCu1-Cu1高出近2倍,实现了高效的PDS 活化和新兴污染物的去除(图4d)。这种位间距效应源于dCu1-Cu1在5~6 Å 时,PDS 的吸附发生改变,形成Cu1-Cu1位点上的双位点吸附结构,显著增强了界面电荷转移,促进了PDS的活化(图4e)。
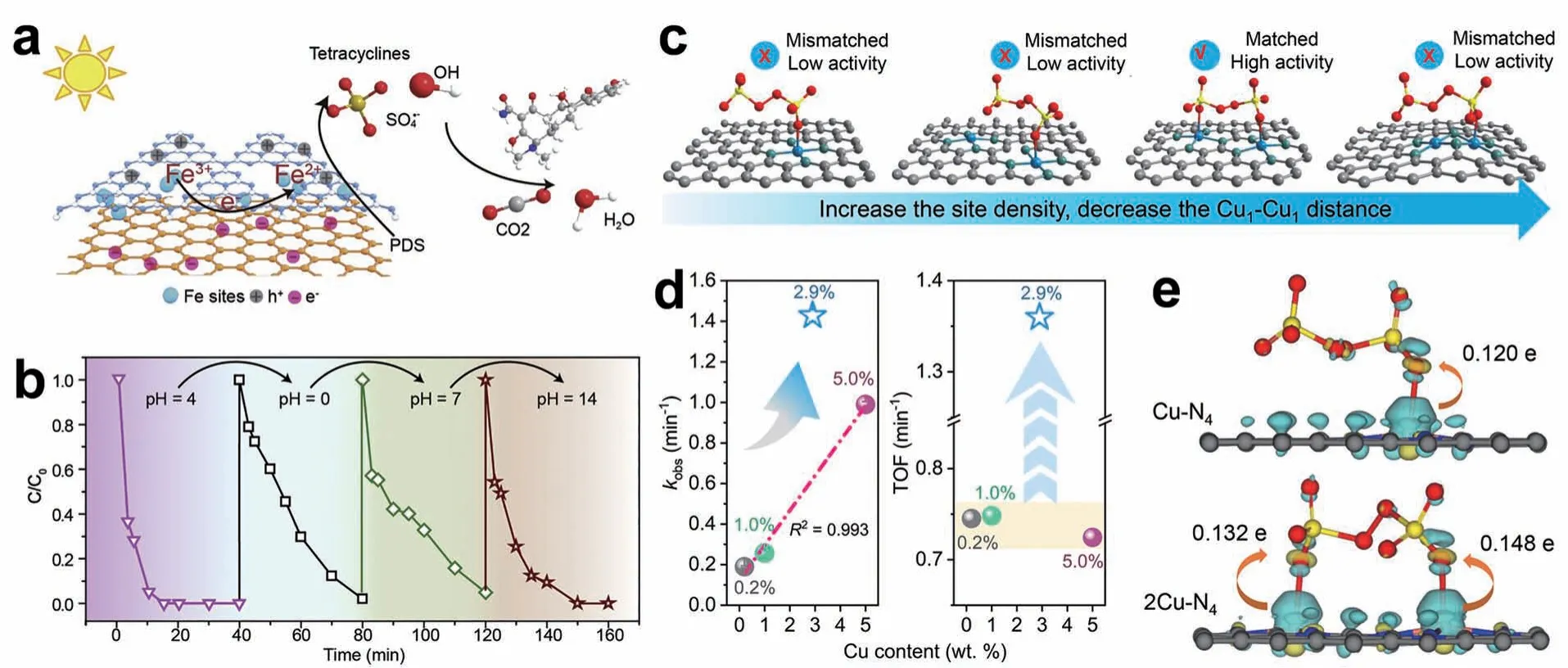
图4 (a) C3N4-Fe-rGO/PDS 类芬顿反应机理的示意图[4];(b)以C3N4-Fe-rGO 为催化剂,考察了不同pH 值条件下溶液的循环降解性能[4];(c) Cu1-Cu1距离对PDS 吸附和活化的影响[41];(d)不同Cu 位密度的Cu1/NG 活化PDS 去除BPA 的一级速率常数和TOF[41];(e) PDS 吸附在2Cu-N4和Cu-N4的电子密度差及相应的电荷转移。黄色和青色轮廓分别代表电子积累和缺失[41]Fig. 4 (a) Scheme diagram proposed for the Fenton-like reaction mechanism of C3N4-Fe-rGO/PDS[4];(b) The degradation performance during cycles of solution with different pH conditions,with C3N4-Fe-rGO as the catalyst[4];(c) Depiction of Cu1-Cu1 distance manipulation for PDS adsorption and activation[41];(d) First-order rate constants and TOFs of BPA removal by Cu1/NG with different Cu site densities activating PDS[41];(e) Electron density difference for PDS adsorption on 2Cu-N4 and Cu-N4 and the corresponding charge transfer.Yellow and cyan contours stand for electron accumulation and deletion,respectively[41]
虽然PDS 和亚硫酸盐的氧化性不如PMS,但由于其低成本和低pH 值对水体的影响而备受关注。此外,通过合理的催化剂调控可以优化PDS 的活化性能,达到能与PMS 体系相近的催化活性。但PDS 体系与PMS 同样存在着出水中SO2-4浓度过高的问题。
2.3 过氧化氢(H2O2)
H2O2以其低成本、环保和无污染的优点在环境修复中受到广泛的青睐,是最有实际应用意义的氧化剂,已经广泛地用于污水的深度处理领域[42-43]。Cui 等[44]证明了SACs 在AOPs 领域实际应用的可能性(图5a)。类芬顿反应中活性金属位点对•OH的淬灭作用是限制其催化活性的原因之一,而SACs中活性位点均匀分散可以解决这一问题。连续反应200 h 后,染料的去除率仍为100%(图5b)。他们设计的芬顿过滤器将催化剂装载在碳纤维表面,无需考虑催化剂回收的问题。催化剂失活后,只需更换滤芯,反应器就能继续运行。此外,原位连续生产低成本的H2O2也是该研究的一大优势,这可以解决H2O2的储存与运输问题,为偏远地区废水处理提供理论和实践支撑。Fu等[45]合成了含有轴向五配位构型(Fe-N5)的类芬顿SACs,极大地改变了铁原子的电子状态,降低了H2O2产生•OH 的激活能垒(图5c 和5d)。此外,富集的吡啶N 作为污染物吸附点缩短了•OH 的扩散距离,与Fe-N5位点建立了双位点反应机制。因此,Fe-N5催化剂对多种污染物的催化氧化表现出高效的类芬顿活性(图5e)。有趣的是,An等[46]发现在超小团簇和单原子铁位点共存的H2O2活化体系中,亚甲基蓝(MB)的去除效率高于纯铁单原子体系。催化性能(以每个催化剂中铁的重量为标准)随着团簇/单原子比率的增加而增加,这表明团簇的催化活性可能优于单原子。
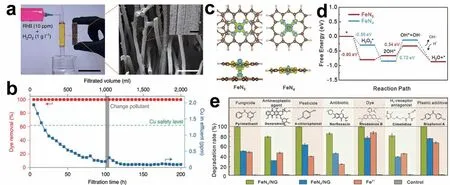
图5 (a)芬顿过滤器的图像及过滤介质的SEM 图像。比例尺:100 μm。内页:涂在碳纤维表面的Cu-C3N4催化剂放大的SEM 图像。比例尺:5 μm[44];(b)染料去除率和废水中的铜浓度与过滤时间的关系[44];(c) FeN5和FeN4模型的差分电荷图。黄色和天蓝色区域分别代表电子积累和电子消耗[45];(d) FeN5和FeN4模型反应过程能垒图[45];(e) FeN5/NG+H2O2、FeN4/NG+H2O2和常规均相Fenton 对不同有机污染物的降解[45]Fig.5 (a) Photo and SEM image of the filter medium.Scale bar:100 μm.Inset,magnified SEM image showing the Cu-C3N4 catalyst coated on the surface of a carbon fibre.Scale bar:5 μm[44];(b) Dye removal and Cu concentration in effluent as functions of filtration time[44];(c) Charge density differences of FeN5 and FeN4 models.The yellow and skyblue regions represent electron accumulation and electron depletion,respectively[45];(d) Energy diagram of the reaction process for FeN5 and FeN4 models[45];(e) Degradation of select organic pollutants in FeN4/NG+H2O2,FeN5/NG+H2O2,conventional homogeneous Fenton (Fe2++H2O2) and control (H2O2)systems[45]
芬顿反应是降解有机污染物最有效的AOPs 之一,可从H2O2中产生高活性的•OH。此外,H2O2具有成本低、产品绿色的特点,在高级氧化领域备受青睐[47-48]。与其它氧化剂相比,H2O2成本低,产品的环保不会造成二次污染问题,是工程应用的首选。然而,SACs 催化H2O2的研究相对较少,主要原因是单原子催化剂很难在H2O2的活化中表现出良好的性能。且H2O2体系通常存在pH值的限制,仅在较低pH值时能发挥较好的催化效果。因此,未来应深入研究H2O2的活化机制,解决SACs难以活化H2O2的关键问题。相信结合SACs和H2O2的优势,有望在实际污水处理中表现出更强大的优势。
2.4 其它氧化剂
除了上述氧化剂外,SACs 在过氧乙酸(PAA)、高碘酸盐)以及臭氧的催化中也表现出优异的性能。Chen 等[49]设计并合成了单原子铁锚定富氮g-C3N4纳米管(FeCNs),并选择PAA 作为类芬顿反应的氧化剂。构建的多相体系在3.0~9.0 的pH 值范围内对多种有机污染物的降解能力增强,表现出很高且稳定的催化活性,比同等数量的原始g-C3N4的催化活性高出75 倍(图6a)。18O 同位素标记技术、探针方法和理论计算表明,PAA 的高效催化活性依赖于高价铁氧与PAA 生成的有机自由基的耦合(图6b 和6c)。Long 等[50]展示了使用SACs 进行分析的第1 个实例。N-rGO-CoSA 在不产生自由基的情况下,有效地活化并在pH 值为3.2~8.9 的范围内降解有机污染物,而Co2+和Co3O4不能驱动活化,Co-N 配位位点表现出较高的活化效率(图6d 和6e)。导电石墨烯基体减少了这些氧化反应的污染物/电子传递距离/阻力,并通过与金属中心协同工作提高了活化能力。电化学实验表明,N-rGO-CoSA/体系通过电子转移的非自由基过程而非分解有机污染物,其中N-rGO-CoSA/亚稳配合物是主要的氧化物质。该工作为设计高效的活化剂以选择性氧化有机污染物开辟了新的途径(图6f)。
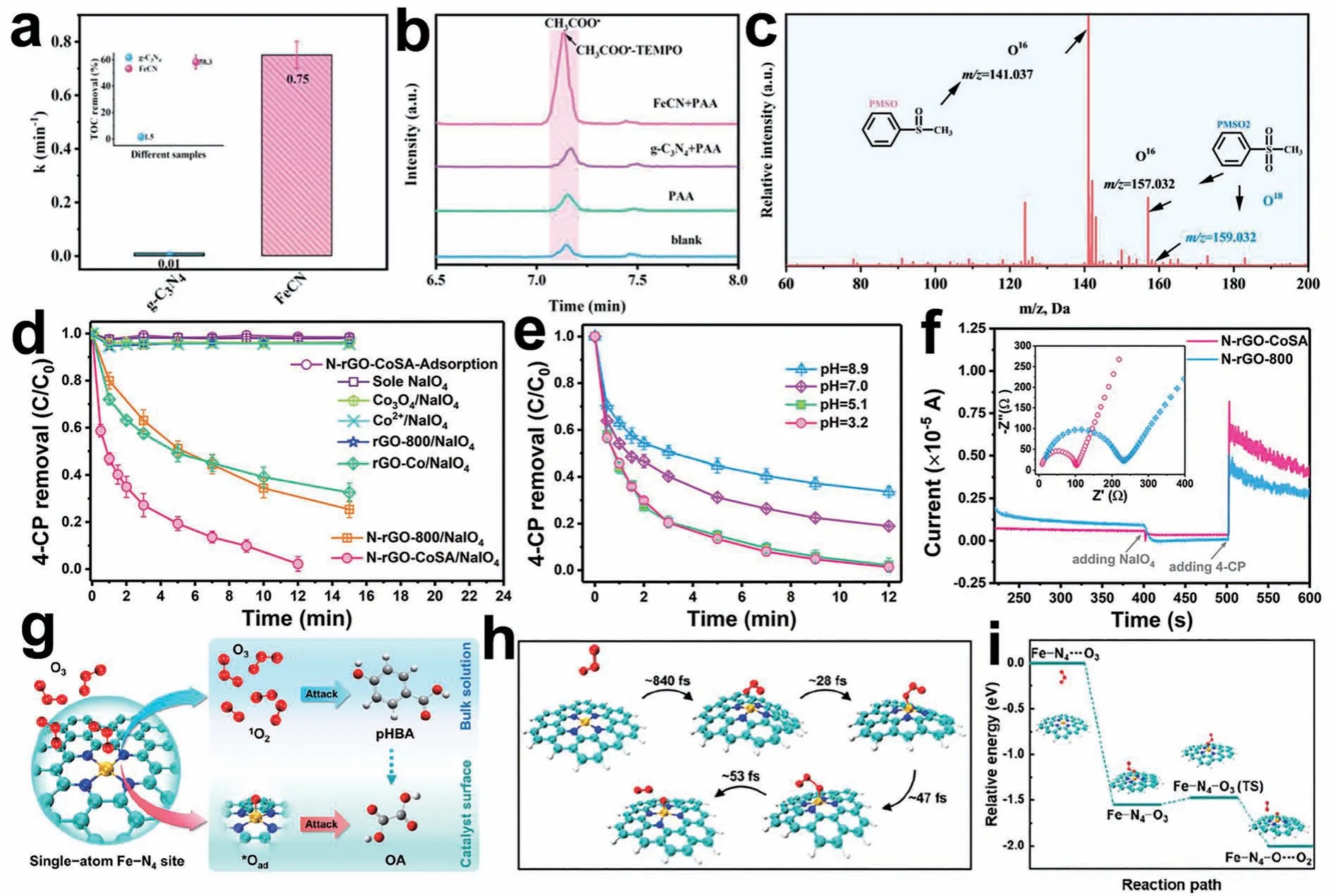
图6 (a) g-C3N4和FeCN的表观速率常数比较(插图:总有机碳(TOC)结果)[49];(b) CH3COO•-TEMPO全扫描色谱图[49];(c) FeCN+PAA 体系中18O 标记或未标记PMSO2的质谱分析[49];(d)不同活化剂活化 降解4-CP[50];(e)溶液初始pH 值对N-rGO-CoSA/ 体系中4-CP 降解的影响[50];(f)在N-rGO-CoSA 粉末包覆的工作电极上依次加入 和4-CP 后的电流响应。插图显示了N-rGO-CoSA 和N-rGO-800 电极在电解质溶液中的EIS 谱图[50];(g) Fe5-NC/O3体系中底物依赖性示意图[51];(h) AIMD 模拟的动态过程[51];(i) DFT 计算的O3与Fe-N4位点相互作用的相对能量分布[51]Fig. 6 (a) Comparison between the apparent rate constants of g-C3N4 and FeCN (inset:TOC result)[49];(b) Full-scan chromatogram of CH3COO•-TEMPO[49];(c) Mass spectral analyses of the 18O-labeled or unlabeled PMSO2 generated in the FeCN+PAA system[49];(d) Degradation of 4-CP by activated using different activators[50];(e) Initial solution pH on the degradation of 4-CP in the N-rGO-CoSA system[50];(f) Current response after the sequential addition of and 4-CP at the working electrode coated with the N-rGO-CoSA powder.The inset shows the EIS profiles of the N-rGO-CoSA and N-rGO-800 electrodes in the electrolyte solution[50];(g) Substrate dependence diagram in Fe5-NC/O3 system[51];(h) Dynamic processes simulated by AIMD[51];(i) Relative energy profile calculated by DFT in the interaction of O3 and the Fe-N4 site[51]
臭氧作为一种强氧化性,无二次污染的绿色氧化剂也受到了广泛地关注。Ren 等[51]提出了一种基于单原子铁催化剂的非均相催化氧化新工艺,其中固定在碳骨架上的Fe-N4位点在降解草酸(OA)和对羟基苯甲酸(pHBA)以及深度处理垃圾渗滤液二级出水方面表现出出色的催化臭氧化活性和稳定性。与传统的自由基氧化不同,他们发现了基于表面吸附原子氧(*Oad)和1O2的非自由基氧化途径。OA被吸附在催化剂表面,主要被*Oad降解,而pHBA主要被本体溶液中的臭氧和1O2去除(图6g)。密度泛函理论计算和分子动力学模拟表明,臭氧末端的一个氧原子优先与Fe-N4的中心铁原子结合,随后诱导催化剂表面附近的O—O键断裂,生成*Oad和1O2(图6h和6i)。此外,Guo等[52]介绍了一种由单Mn原子锚定在石墨氮化碳上的非均相催化剂,通过改变反应途径,有效地克服了酸性条件下O3/H2O2体系反应速率常数有限的弊端,从而显著促进了酸溶液中•OH 的生成。实验与理论相结合的研究表明,Mn-N4是催化活性位点。他们发现了一种独特的H2O2活化生成的催化途径,摆脱了传统臭氧反应中作为必需引发剂的限制。
3 类芬顿反应中SACs的实际应用测试
催化剂的稳定性和可重复使用性是关系到其实际应用的重要因素[53]。在最初的研究中,SACs 中较低的金属负载被认为是导致其稳定性较差的原因。随着对SACs的深入研究,研究人员对SACs和纳米颗粒催化剂的活性有了新的认识。在合成SACs 的过程中,为了避免金属的聚集,通常会选择N 或S等配位元素含量高的载体,这些载体可以发挥“爪子”的作用,有效地结合金属离子。因此,SACs 实际上拥有非常稳定的配位结构,通过柱状或膜状的芬顿反应器实现了SACs在AOPs中稳定性的提高[54-60]。
3.1 柱状反应器
2020 年,Cui 等[44]设计了SACs 芬顿过滤器开创了单原子催化剂实际应用探索的先河。芬顿过滤器连续运行200 h 后(过滤了2 L 废水),染料去除率保持在100%。这是SACs 的第一个设备级演示,以验证其实际应用潜力。Zuo 等[55]利用垃圾渗滤液(化学需氧量(COD)=1759 mg/L)测试了Cu-SA 和H2O2/PDS/PMS 体系的实际应用能力,处理1 h 后,COD 去除率由30%提高到60%。在较高的流量下,连续运行50 d 后,Fenton 反应器的BPA 去除率仍高达100%(反应条件:污染物浓度为0.1 mmol/L,氧化剂浓度为1 mmol/L,流速为10 mL/min)(图7a)。此外,Liang 等[56]将制造的Co-SAC 用于连续流反应器中。在连续操作168 h 的情况下,双酚A 的去除率保持在100%,而且Co 离子浸出率很低,这表明Co-SAC 具有良好的催化活性和耐久性。并且,通过比较Co-SAC 催化反应后与新鲜催化剂的XRD 图谱和HAADF-STEM 图像,进一步验证了Co-SAC 的良好稳定性。
3.2 膜状反应器
与柱式反应器相比,膜式反应器由于其较高的水流量和较短的水力停留时间[57-59],有更大的挑战。Zhang 等[59]通过Fe-CNW3 包覆膜过滤器连续流去除BPA 的实验来评价该体系的实际应用性能。在连续运行170 h 后,去离子水中BPA 的去除率保持在90%以上(图7b)。Ma 等[60]设计了用Cu 单原子催化剂(Cu1-M)和Cu 纳米颗粒催化剂(Cunano-M)支持的催化膜(图7c)。Cu1-M 在300 min 的连续过滤中表现出稳定的对乙酰氨基酚去除率,而催化活性只有轻微下降。相比之下,Cunano-M 的水净化性能随着时间的推移而急剧下降,300 min 后膜完全失去活性(图7d)。Cu1-M 的稳定性归功于富含电子的反应载体(S 基),它保持了铜原子的低氧化状态以进行催化作用。然而,Cunano-M 中没有足够的电子可用于Cu 的还原。Wang 等[54]在设计的膜反应器中使用Co-N3O1,发现水流在PVDF/Co 中的停留时间只有312 ms。而且在PVDF/Co/PMS 体系中,CIP 的去除率在10 h 内保持在97.5%,表明Co-N3O1对PMS 的活化具有很高的稳定性和活性。目前,很多连续流类的实际应用测试并没有提到单位催化剂的废水处理能力。例如,如果无限制增加催化剂的量,理论上讲可以无限制增加该反应器的污水处理能力。因此,仅仅依靠去除率和废水处理量是不合理的。
所有这些关于SACs 实际应用潜力的设备级演示均表明了SACs 的高耐久性和活性。尽管纳米级催化剂也具有良好的可回收性,但它们的稳定性可能主要靠大量金属离子的浸出来维持,而不像SACs那样依靠强大的金属-载体相互作用。催化剂的活性、稳定性和价格一直是限制非均相催化剂实际应用的重要因素。随着SACs 的出现和深入研究,相信这些问题最终会被克服,越来越多的SACs 将被用于实际污染物的处理。
4 结论与展望
近年来,非均相催化剂在AOPs 中的应用取得了很大进展。催化性能对金属中心的尺寸和结构很敏感。金属活性中心尺度的改变不仅会影响催化活性,而且会使催化剂在催化选择性和稳定性方面表现出相当大的差异。具有原子活性位点的SACs 有利于探索结构-活性关系,可以进一步开发更先进的非均相催化剂。本文总结了SACs 的合成调控方法。此外,还详细总结了SACs 在类芬顿反应中的催化表现和实际应用潜力。最后,对SACs 在类芬顿反应中遇到的问题和未来的发展方向做了总结和展望。受益于超高的反应性、选择性和稳定性,SACs 在实际商业应用中显示出巨大的潜力。尽管在活性中心的尺度调控、表征和机理探索方面已经取得了突破性进展,但SACs 在AOPs 领域的应用仍处于起步阶段。一些基本问题仍有待解决,以便为先进的类芬顿催化剂的未来发展铺平道路。1)开发模型催化剂。活性位点本征特性的不同是导致不同催化机制的最关键原因,因此研究人员开发了不同金属尺寸或配位环境的催化剂进行比较。控制不同尺寸或配位环境需要改变合成条件,如通过掺杂杂原子改变配位微环境,通过控制煅烧温度调整不同N 物种的比例。然而,合成条件的改变也会对底物的特性产生影响,这就导致了对不同配位环境的不精确的比较。模型催化剂的开发可以比较特定尺度或配位之间的差异,有效避免了其它因素的影响。这对于精确确定反应机理,研究不同配位和尺度之间的关系具有重要意义。2)制备M-M 配位的SACs。在传统的SACs 中,金属原子的低负载导致单元位点之间的距离较大,缺乏间距协同作用。目前,关于尺度对催化活性的影响,结论并不严谨。因为在以前的研究中,SACs 和纳米级催化剂的不同性能主要是由于活性位点密度的不同,而没有关注它们电子特性的差异。具有M-M 配位的SACs 的缺乏是限制结构-活性关系研究的关键。经过10 多年的发展,SACs 的制备方法已经相对成熟。然而,目前SACs 的制备仍集中在保证金属原子的充分分散、提高金属负载量以及合成方法的多样性上。最近的研究发现,M-M 配位环境的独特电子结构和令人满意的催化活性。然而,很少有人系统地比较孤立金属、双金属、甚至多金属配位的内在特性和催化活性。研究人员已经发现,负载量的变化可以影响金属位点的距离,从而调节金属尺度的大小。尽管如此,这些方法大多依赖于特定的基质或配位环境,并不具有普遍性。有必要开发通用的方法来精确控制金属的尺寸。对结构-活性关系的研究对于理解催化机制和开发先进的类芬顿催化剂至关重要。3)忽略了载体的作用。碳材料或其它金属材料在AOPs 领域具有突出的作用,特别是对PMS 的活化作用。然而,许多研究只关注活性金属中心的作用,而忽略了载体对催化活性的影响。在研究活性金属中心的作用之前,应排除载体的作用。4)应关注污染物和氧化剂的影响。越来越多的研究表明,催化剂不是决定污染物降解机制的唯一因素。不同的污染物类型和氧化剂浓度也会对反应机制产生重要影响。例如,污染物的半波电位和电子富集度也直接影响其在催化剂上的吸附和与活性氧物种(ROS)的反应,PMS 的负载也会影响自由基与非自由基的比例[61]。此外,非均相催化剂对污染物的去除包括吸附和降解2 个步骤,有机分子在催化剂上的吸附也会引起催化剂的电子结构和轨道特性的变化。因此,催化部位的氧化剂吸附和活化行为也会发生变化。从这个角度看,在探索活性物种时,不仅要考虑催化剂和氧化剂的作用,还要考虑污染物的作用。5)加强新的表征技术的应用。 活性位点的确定和反应机理的探索是理论研究的重要方面。表征方法的缺乏也是限制结构-活性关系研究的一个重要原因。例如,SACs 中的金属-金属相互作用是现有表征方法难以发现的;团簇大小和局部配位构型等变量对表征方法构成了巨大挑战;不发达的表征方法会误导DFT 计算,使计算结果不准确。因此,开发新的表征方法或原位表征技术可以促进催化机制的研究。此外,基于机器学习的理论模拟也应得到更多关注。
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
当代陕西(2019年6期)2019-04-17
中国资源综合利用(2017年2期)2018-01-22
中国资源综合利用(2017年2期)2018-01-22
环境科技(2016年3期)2016-11-08
中国资源综合利用(2016年11期)2016-01-22
淮南师范学院学报(2015年3期)2015-03-22
河北科技大学学报(2015年5期)2015-03-11
无机化学学报(2014年4期)2014-02-28
无机化学学报(2014年4期)2014-02-28
