
过渡金属催化羰基化反应的最新研究进展
2024-05-15 09:06徐山梅高帆孙国庆李明权正军
西北师范大学学报(自然科学版) 2024年3期
关键词:一氧化碳
徐山梅 高帆 孙国庆 李明 权正军


DOI:10.16783/j.cnki.nwnuz.2024.03.002
收稿日期:2024-02-28;修改稿收到日期:2024-04-12
基金项目:国家自然科学基金资助项目(22061038,22067018);甘肃省博士后基金资助项目(00245);西北师范大学学科建设基金资助项目(202103101413,202303101410)
作者简介:徐山梅(1995—),女,甘肃庆阳人,硕士研究生.主要研究方向为Pd催化羰基化反应.
E-mail:1844973622@qq.com
*通信联系人,男,教授,博士,博士研究生导师.主要研究方向为杂环化合物的合成、催化有机合成、含磷有机化合物的合成和富硫聚合物制备及其应用等.
E-mail:quanzhengjun@hotmail.com
摘要:过渡金属催化羰基化反应是制备羰基化合物最直接的方法之一,近年来得到了迅速的发展.利用活性高且廉价的CO气体作羰基源,通过过渡金属催化的方法实现羰基化,是现代有机合成中构建含羰基化合物的有效方法.基于反应原料和产品氧化还原价态的改变等不同方式,将过渡金属催化羰基化反应分为四种主要类型:杂核亲核试剂的羰基化反应,碳亲核试剂的羰基化反应,碳亲电试剂的羰基化反应和还原偶联的羰基化反应,分别综述了各类型羰基化反应的最新研究进展.
关键词:过渡金属催化;一氧化碳(CO);羰基化反应;交叉偶联;还原偶联
中图分类号:O 621.3 文献标志码:A 文章编号:1001-988Ⅹ(2024)03-0005-20
Recent progress in transition metal-catalyzed
carbonylation reactions
XU Shan-mei,GAO Fan,SUN Guo-qing,LI Ming,QUAN Zheng-jun
(Gansu International Scientific and Technological Cooperation Base of Water-Retention Chemical Functional Materials,
College of Chemistry and Chemical Engineering,Northwest Normal University,Lanzhou 730070,Gansu,China)
Abstract:Transition metal catalyzed carbonylation reaction is one of the most direct methods for the preparation of carbonyl compounds,which has been developed rapidly in recent years.The use of highly active and inexpensive CO gas as a carbonyl source to realize carbonylation via transition metal-catalyzed methods is the most effective strategy for atomically economical construction of carbonyl compounds in modern organic synthesis.Herein,the transition metal-catalyzed carbonylation reactions reported in recent years are classified as into four categories based on the oxidation-reduction valence state change of the reactant material and product,that is,carbonylation reaction of heteronuclear nucleophiles,carbonylation of carbon nucleophiles,carbonylation reaction of carbon electrophiles,and reductive coupling carbonylation.
Key words:transition metal catalysis;carbon monoxide(CO);carbonylation reaction;cross coupling;reduction coupling
羰基骨架廣泛存在于天然产物及药物分子中[1],它们是构建C-C和C-X键的重要官能团.因此高效构建羰基骨架在有机合成领域具有重大意义.CO是一种自然界中广泛存在的可再生资源[2],也是羰基化反应中最常用的羰基源.
多组分反应(MCR)是一种快速增加分子复杂性的直接方法,其经由多个反应物通过串联方式引入多个活性官能团,体现了MCR的原子经济性[3].使用CO气体或CO的替代物作为羰基源,能够从简单的起始原料有效地构建含羰基化合物[4].与经典的多组分反应相比,羰基化反应具有更高的原子经济性,但同时也具有更大的挑战性[5-6].1938年,Otto Roelen小组在研究费托反应时发现了氢甲酰化反应,这也是第一个过渡金属催化的羰基化反应,在烯烃的碳碳双键上引入一个醛基(CHO)和一个氢原子,该工艺已被广泛应用于烯烃制醛的工业中.1970年,Monsanto公司开发了以铑/碘化物为催化剂的甲醇低压羰基化制醋酸的工艺,取代了1960年由BASF公司首创的碘化钴催化的高压工艺.BP公司于1996年推出了Cativa工艺技术专利,采用基于铱催化的新体系改进了传统甲醇羰基化的过程,相比于铑催化体系,铱催化体系具有更高的活性和选择性,且体系副产物少.因此,后续70%的工业乙酸生产中都采用Cativa的甲醇羰基化(图1)[7].
carbonylation reaction
20世纪70年代,自Heck首次报道Pd催化羰基化反应构建酰胺的开创性工作以来[8],过渡金属催化羰基化反应已成为有机合成领域中的重要研究内容,备受研究者们的关注与重视[9].在合成醛、酮、羧酸及其衍生物方面具有广泛的应用[10],其不仅可以用于合成天然产物、药物分子等复杂有机分子[11-12],还可以应用于材料科学领域,如聚合物合成、功能材料制备等方面[13].
在过渡金属催化下,使用CO作为羰基源的羰基化反应还面临很多挑战.例如,CO在常温常压下是一种无色无味的剧毒气体,这使得它的使用和储存存在一定的安全问题.利用廉价而丰富的CO作为C1源,通过过渡金属催化(Pd,Rh,Cu,Ni,Fe,Mn)将一个或多个CO单元结合到有机化合物的羰基化反应已经成为一个快速构建羰基化合物的有效策略[14-15].本文综述了近年来过渡金属催化的不同类型的多组分羰基化反应的研究进展.
1 杂核亲核试剂的羰基化反应
1.1 Pd催化氮、氧亲核试剂参与的羰基化反应
2019年,Zhang等[16]报道了一种通过乙烯基三氟甲磺酸酯和亲核试剂(氢气、醇、胺、苯酚)的羰基化合成α,β-不饱和醛、酯和酰胺的有效方案(图2),该合成的关键是使用含有吡啶基取代dtbpx型配体络合的钯催化剂.
高活性卤化物(碘化物或溴化物)作为常见的偶联试剂,被广泛应用于羰基化反应中.2022年,Wang等[17]首次报道了反应性较低的芳基氯化物与脂肪族伯胺、仲胺以及芳香胺,在醋酸钯和Xantphos的催化体系中直接羰基化偶联反应(图3),其中CsCl是实现该反应的关键添加剂.这种催化体系能够使多种芳基氯化物与芳香胺、烷基胺高效转化为酰胺化合物,表现出良好的官能团相容性,成功解决了低活性氯化物难发生羰基化反应的问题.
2023年,Bao等[18]开发了一种钯催化溴代乙腈与亲核试剂的羰基化反应(图4),构建了一系列α-氰基取代的羧酸衍生物.在温和条件下,使用较低催化量的钯和配体进行克级放大实验,并以优异的产率获得了目标产物.此外,这种转化也可以在常压CO气氛下进行,为多种药物前体的制备提供有效途径.
Pd催化Heck型羰基化反应可以快速构建具有羰基基团的苯并杂环衍生物,为生物医药合成提供了一定的参考.2019年,Wang等[19]开发了一种新颖的钯催化N-(2-溴苯甲酰基)吲哚去芳构化羰基化反应(图5).该催化系统由市售的Pd(OAc)2和dppp组成,并使用醇和苯胺作为亲核试剂,以中等至良好的收率提供所需目标产品.值得注意的是,该转化是羰基化脱芳构化的第一个例子.
2020年,Chen等[20]报道了单齿亚磷酰胺配体Xida-Phos辅助Pd催化的芳基卤代试剂(N-芳基丙烯酰胺)和各种亲核试剂(芳基硼酸、 苯胺和醇)与CO的多米诺Heck型羰基化反应(图6).该反应显示出优异的反应性和对映选择性,具有良好的官能团耐受性,提供了直接获得各种具有β-羰基取代的季碳立体中心吲哚酮衍生物,为合成各种具有不对称生物活性的六氢吡咯并吲哚及其二聚生物碱提供了一種简便且互补的方法.
同年,Yuan等[21]报道了钯催化的Heck型去对称化构建手性双环[3.2.1]辛烷羰基化反应(图7).醇、酚和胺均可被用作该体系的亲核偶联试剂,以此构建多官能化的手性双环[3.2.1]辛烷,所得产物具有一个季碳和两个叔碳手性中心,并具有高度的非对映和对映选择性.该研究代表了不对称串联-Heck-羰基化反应和内部烯烃的对映选择性双官能团化的重要进展.反应机理如图7所示,Pd(0)催化剂9与芳基碘代试剂7进行氧化加成得到络合物10,随后发生分子内Heck环化生成烷基钯中间体11,最终CO的配位插入及后续的亲核进攻、还原消除过程得到目标产物8.
2021年,Hu等[22]采用与Chen等[20]类似的机理,开发了第一例钯催化卤代物参与的串联双羰化-氨基化反应(图8),通过钯催化Heck型异环化反应中间体,双CO分子选择性地插入σ-烷基钯中间体及后续的烷基胺作为亲核试剂提供了一系列双羰基氨化产物.合成的带有多个活性位点的α-酮酰胺可作为经典反应的多功能中间体,也可以被应用于天然产物和药物分子骨架的修饰中.
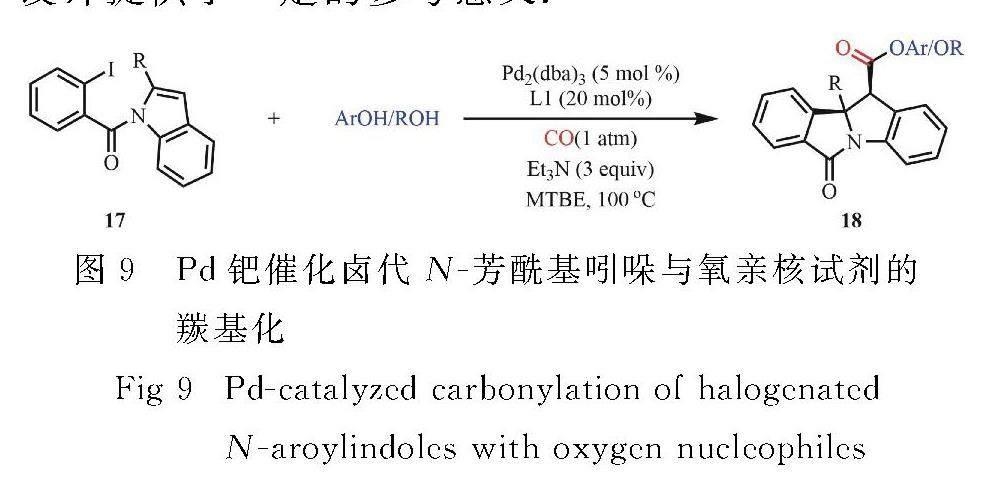
2022年,Li等[23]报道了钯催化的N-芳酰基吲哚的对映选择性去芳构化、羰基化反应(图9).该方法通过一锅法实现了带有连续C2-氮杂五元和C3季碳中心的稠杂环衍生物的构建.该反应底物范围适用性强,为后续不对称羰基化反应的进一步设计提供了一定的参考意义.
同年,Chen等[24]报道了钯催化的取代环丁酮与CO的不对称C-C键活化羰基化反应(图10).该方案反应产率高,对映选择性高,具有良好的官能团耐受性,为C3位含季碳立体中心的手性茚满酮的合成提供了一种有效的方法.
2022年,Li等[25]通过使用烯烃取代的环丙烷开环策略,在过渡金属钯催化下,实现了氨甲酰基取代茚酮的制备(图11).一系列结构复杂的胺亲核试剂(包括氨基酸衍生物和几种药物)等都能够被应用到该反应体系中,其可能的反应机理如图11所示.起初Pd(0)与27的氧化加成得到芳基Pd络合物29,之后第一分子CO配位并插入到芳基Pd生成酰基Pd络合物30,30与相邻的双键配位加成得到中间体31,31经β-碳消除生成烷基Pd中间体32,随后经胺化、配体交换后插入第二分子CO,得到中间体33,最后经还原消除得到产物和Pd(0)物种.
1,4-Pd迁移是一种实现远程C-H活化的有
效途径.2020年,Cˇarny等[26]通过1,4-Pd迁移实
现了远程C(sp3)-H键的氨基、烷氧基羰基化反应(图12),以中等至良好的产率构建了一系列含有β-季碳中心的羧酸衍生物.
2023年,Li等[27]报道了Pd催化下邻溴二苯乙烯、CO和亲核试剂的区域选择性三组分羰基化偶联反应(图13),用以合成多取代的α,β-不饱和羰基化合物,其中涉及钯催化的1,4-Pd迁移策略实现了对C(sp2)-H活化、羰基化过程.该策略的显著特征包括使用标准大气压的CO、优异的化学选择性以及良好的官能团耐受性.
通过DFT计算了CO迁移插入和亲核试剂进攻的先后顺序,并提出了可能的反应机理(图14).首先,原位生成的Pd0物种39与邻溴二苯乙烯37发生氧化加成生成中间体40.随后通过阴离子交换得到物种41,再通过协同金属去质子化(CMD)机制进行C(sp2)-H活化,得到五元环钯中间体42.42经质子化开环得到烯基钯中间体43,然后中间体43经历连续的CO迁移插入、亲核试剂进攻得到中间体45,最后还原消除生成目标产物和再次参与催化循环的Pd0物种39.
双官能团化的羰基化反应是一类快速构建复杂羰基化合物的有效方法.2016年,Wang等[28]报道了一种炔烃的双官能团化羰基化反应(图15).通过使用Pd催化实现了二氟烷基化、羰基化,构建了一系列含二氟烷基的α,β-不饱和羧酸衍生物,其中炔烃和亲核试剂的广泛适用性证实了该合成方法的研究价值.机理研究表明,二氟烷基自由基的参与是该反应转化的关键:首先二氟碘乙酸乙酯与Pd0之间通过单电子转移途径分别得到PdⅠ物种和二氟烷基自由基48,随后48与苯乙炔加成得到
中间体49,再与PdⅠ物种结合得到PdⅡ物种50,随后CO迁移插入得到酰基PdⅡ物种51,最后亲核试剂在碱作用下与酰基PdⅡ物种51发生亲核取代,再还原消除得到最终的目标产物46.
2021年,Zhang等[29]报道了钯催化全氟烷基卤化物、非活化烯烃、胺和CO的双官能团化的羰基化反应(图16),这是一种构建全氟烷基酰胺衍生物的新方法.各种胺包括磺酰胺和肼在内的多种亲核试剂都是该催化体系的有效亲核偶联试剂,生成了各种具有良好官能团耐受性和高化学选择性的β-全氟酰胺化合物52.
2022年,Cheng等[30]报道了钯催化芳基碘化物与烯基异氰、CO和胺的四组分串联羰基化反应(图17).不同于上述反应,该反应是通过氧化加成、分子内环化和还原消除等过程进行的.该反应在一锅法串联过程中一步形成4个化学键,合成了一系列乙酰胺取代的五至七元环酮骨架.此外,通过原位或反应后将氨基NH环化为酮亚胺部分,形成了一个额外的C—N双键,为构建吡咯并稠杂环提供了一种简便的途径.该反应可能的机理为(图17):首先芳基碘化物与Pd(0)发生氧化加成得到芳基PdⅡ物种55,随后中间体55与异氰化合物53配位迁移插入生成亚氨基PdⅡ物种56,该中间体通过分子内环化加成形成烷基PdⅡ物种57,随后CO配位、插入形成酰基PdⅡ中间体58,58被胺亲核进攻得到中间体59.最后,59还原消除得到最终产物54并同时生成Pd0活性物种参与下一个催化循环.
1.2 Rh催化氧、硫亲核试剂参与的羰基化反应
2020年,Ai等[31]报道了一种在低压CO气氛下,铑催化的烷基卤化物与酚类化合物的羰基化反应(图18).这种稳定的催化剂体系能够使用酚类作为羰基偶联的亲核试剂,同时表现出高官能团耐受性和良好的化学选择性.具有大空间位阻基团或多个反应位点的底物也可以选择性地转化为所需的目标产物,产率较高.
通过对照实验提出了可能的反应机理(图19):首先活性RhⅠ物种60和烷基卤化物通过氧化加成得到烷基RhⅢ中间体61,这可能是该反应的决速
步骤,随后烷基RhⅢ中间体61与CO配位迁移插
入生成酰基RhⅢ络合物62,在碱作用下,苯酚亲核进攻酰基RhⅢ络合物62得到RhⅢ络合物63,最后,RhⅢ络合物63进行还原消除生成最终的酯类化合物,同时再生成活性RhⅠ络合物60参与下一个催化循环.
由于苯硫酚的高亲核性,使得苯硫酚与烷基卤代物之间的亲核取代反应较容易发生,同时硫醇类化合物对过渡金属具有很强的亲和力,这会导致催化剂失活.因此,过渡金属催化卤代烷与硫醇类化合物的羰基化反应是具有挑战性的研究课题.
2021年,Ai等[32]使用苯硫酚与烷基碘代物在Rh催化下实现了羰基化反应(图20),这种催化体系有效克服了羰基化反应和亲核取代反应之间的竞争反应,选择性地得到了羰基产物.
2022年,Wang等[33]首次报道了铑催化的烷基氯化物与脂肪醇或酚类化合物的选择性羰基化偶联酯化反应(图21).添加剂碘化钠可以在原位发生卤素交换,从而提供更具活性的烷基碘化物,也是该反应的关键步骤.在RhⅠ-DPPP催化体系中,以高达95%的分离产率制备了多种酯类化合物.该催化体系还用于氯甲烷和二氯甲烷来合成与工业相关的乙酸酯类衍生物.
1.3 Cu催化氮、氧亲核试剂参与的羰基化反应
2019年,Chen等[34]报道了一种在In或InI存在下,通过Cu和In双金属介导作用实现非活化烷基碘化物和醇类的烷氧基羰基化反应(图22),顺利合成了一系列烷基酯类化合物.Cu/In/CO在该羰基化过程中起协同作用,该反应适用于伯、仲甚至叔烷基酯的制備.
同年,Lu等[35]报道了一种不含光敏剂的自由基羰基化反应(图23).在可见光诱导下,Cu催化剂与环丁酮肟酯通过单电子转移方式产生亚胺自由基,随后自由基开环产生含氰基的烷基自由基,再与胺类化合物在CO中实现了羰基化反应.此方法表现出广泛的底物范围,环丁酮肟酯和脂肪族胺和芳胺均具有高度官能团相容性,提供了温和途径下获得不同取代的氰基烷基化酰胺化合物的方法.
2019年,Yin等[36]报道了Cu催化N-氟磺酰胺的远程C(sp3)-H活化的分子内和分子间的羰基化反应(图24).该反应涉及酰胺基自由基生成、1,5-HAT和羰基化过程,以高产率和良好的区域选择性得到了各种δ-内酰胺.这种1,5-HAT的羰基化反应还可以扩展到与不同醇的分子间过程,为形成多样性酯类化合物提供了一种有效方法.另外,该方法还可以用于N-氟甲酰胺的转化和基于药物塞来昔布衍生物的后期官能团转化.
对此,他们提出了一种可能的反应机理(图25).反应始于[Cu]Ⅰ物种70与N-氟磺酰胺67发生的单电子转移,形成磺酰胺基自由基71和[Cu]Ⅱ物种.然后,磺酰胺基自由基经过分子内的1,5-HAT生成新的碳中心自由基中间体72,72会被CO和[Cu]Ⅱ物种捕获,形成酰基[Cu]Ⅲ物种73.随后,73经历分子内或分子间配体交换,形成中间体74或75.最后中间体74或75进行还原消除,生成最终的δ-内酰胺68或酯类化合物69,其中[Cu]Ⅰ物种70参与下一个催化循环过程.
2020年,Zhang等[37]报道了一种在Cu(OTf)2催化作用下N-氟磺酰胺的分子内环化和醇类化合物分子间羰基化的新策略(图26),合成了一系列N-磺酰基-β-高脯氨酸酯类化合物,为合成潜在的功能化生物活性分子提供了可能性.
随后,Zhang等[38]报道了Cu催化γ,δ-不饱和芳香族肟酯和胺类化合物的Narasaka-Heck环化、羰基化反应(图27),该方法是一类高效制备含吡咯烷酰胺类化合物的新方法.
2023年,Zhang等[39]报道了一种可见光诱导的硫鎓盐开环羰基化反应(图28),其中Cu盐作为光催化剂,有效促进了含硫羧酸衍生物的生成.该方法在温和反应条件下可以精确、选择性地断裂硫鎓盐中的C(sp3)-S键,通过自由基过程可以实现具有良好官能团耐受性和优异化学选择性的广泛底物的转化.值得注意的是,这种自由基型开环羰基化体系可以进一步应用于各种生物活性分子的后期修饰,还能够实现克级放大实验.
从同一底物可控性地生成酰胺和α-酮酰胺是一个具有吸引力的目标.2022年,Zhao等[40]开发了一种新型铜催化卤代物与胺类化合物的单(双)羰基化反应(图29).在不同的Cu催化剂和不同压力的CO气氛中,实现了烷基碘化物可控的单/双羰基化反应构建酰胺和α-酮酰胺.当体系中加入催化剂量的Co2(CO)8时,烷基溴化物发生高选择性双羰化反应,以中等至良好产率获得了唯一产物α-酮酰胺.
烯烃的双官能团化是一种从易得的起始原料合成复杂化合物的有效策略.2021年,Wu等[41]开发了一种Cu催化未活化烯烃的三氟甲基化、羰基化反应(图30).从具有优异区域选择性的简单烯烃中,以中等至优异的产率制备了范围广泛的β-三氟甲基化羧酸衍生物.特殊的是,结构最简单的乙烯气体也可直接用于制备β-三氟甲基化的羧酸衍生物82.这也是首次通过Cu催化四组分串联羰基化反应来实现烯烃的三氟甲基化和羰基化的方法.
2022年,Zhang等[42]利用氰化亚铜和BINAP
组成的催化体系(图31),实现了可见光诱导非活
化烯烃的全氟烷基化羰基化反应.该方法实现了全氟烷基碘化物的自由基串联反应,构建了一系列高度官能化的全氟烷基羧酸衍生物.各种烯烃、全氟烷基卤化物和亲核试剂(包括酚类、 醇类和胺类)都适用于该串联羰基化反应,提供了超过70个β-全氟烷基羧酸衍生物的例子,具有良好的官能团耐受性和优异的化学和区域选择性.此外,该方法还成功实现了多种药物分子和生物活性分子参与全氟烷基羰基化反应.
2023年,Zhang等[43]通过可见光诱导的铜催化乙烯气体的三氯甲基化羰基化反应(图32),以市售的CCl4和CO分别作为三氯甲基和羰基源将胺、酚和醇在内的各种亲核试剂快速转化为具有良好官能团耐受性的β-三氯甲基羧酸衍生物84.通过调节CO和乙烯的压力还可得到双乙烯基化的γ-三氯甲基酰胺85.
1.4 其他金属催化氮、氧亲核试剂参与的羰基化反应
2022年,Ai等[44]开发了一种钳形Ru催化体系(图33),有效实现了烷基碘化物与醇类化合物的烷氧基羰基化反应.利用钳形配体的紧密配位,有效防止了Ru催化活性的降低,具有優异的杂环相容性和广泛的底物适用范围.
铁作为一种储量丰富、廉价且毒性较低的过渡金属催化剂具有明显的优势,它成本低,并且具有独特和互补反应模式的潜力,这种反应体系在可持续发展的化学界广受欢迎.
2022年,Ai等[45]报道了关于Fe催化卤代烷烃的烷氧羰基化反应(图34).通过机理研究表明,该反应是由原位生成的Fe2-络合物催化发生.这种低价铁物种通过独特的双电子转移(TET)过程活化烷基溴化物,而通过单电子转移(SET)过程活化烷基碘化物.
烷基溴及具有低亲核性的酰胺和吲哚被认为是羰基化反应特别具有挑战性的底物.2023年,Ai等[46]报道了铁催化非活化卤代烷与胺、酰胺或吲哚的羰基化偶联(图35),以构建酰胺结构单元,并以优异的产率和良好的官能团相容性合成了各种酰胺、酰亚胺和N-酰基吲哚.初步机理研究表明,当使用烷基碘时,羰基化通过自由基途径进行;当烷基溴作为亲电试剂时,通过双电子转移(TET)过程实现羰基化反应.
2023年,Ai等[47]报道了一种钳形锰催化体系,可以有效活化惰性C(sp3)-Cl进行烷氧基羰基化(图36).通过机理探究发现,廉价的Mn催化剂与烷基氯化物通过氧化加成直接活化,以良好的官能团相容性得到了多种取代的羧酸酯衍生物.
2 杂核亲核试剂的羰基化反应
2.1 硼酸类碳亲核试剂
2.1.1 Pd催化硼酸类碳亲核试剂参与的羰基化反应
2016年,Zhao等[48]报道了在一个大气压的CO气氛下,使用过渡金属钯催化,二氟烷基溴化物与芳基硼酸发生交叉偶联的羰基化反应(图37),以中等至优的产率得到目标化合物.该方法的优点是合成简单、底物范围广和官能团兼容性好,所得二氟烷基酮可用作合成含氟化合物的通用结构单元.
用芳基三氟硼酸钾盐代替芳基硼酸时(图38),也能以较高产率得到二氟烷基酮化合物,体现了该类羰基化反应高效率和较高的官能团耐受性.
2019年,Zhang等[49]提出了一种通用且高化学选择性的Pd催化方案,通过乙烯基三氟甲磺酸酯1和芳基硼酸的交叉偶联羰基化反应来合成α,β-不饱和酮(乙烯基酮)87结构(图39).单齿膦配体cataCXiums A对该反应有很好的立体选择性,各种乙烯基酮均能以良好的收率得到目标化合物.
羰基化反应对亲电试剂的种类都有良好的兼容性,如芳基卤化物、芳基三氟甲磺酸酯(来自酚类)和芳基重氮盐(来自苯胺类),但在Suzuki羰基化反应中使用芳香族C—H键作为亲电试剂仍然是一个严峻的挑战.
2023年,Zhang等[50]报到了芳基噻蒽盐与芳基硼酸的钯催化羰基化交叉偶联反应(图40),制备了大量的二芳基酮类化合物,该类化合物在抗肿瘤药物、农用化学和工业合成化学中具有广泛的应用前景,其中芳基噻蒽盐是由芳烃通过位点选择性实现C(sp2)-H噻蒽化得到,巧妙的开发了将惰性的C(sp2)-H键转化为增值分子的温和方法.
2021年,Zhao等[51]在一个大气压的CO气氛下,使用9-BBN化合物90与卤代二氟烷基化合物,以Pd(PPh3)2Cl2为催化剂,XantPhos为配体,K3PO4为碱,二氧六环作溶剂的条件下实现了交叉偶联羰基化反应(图41),其中,加入当量水的作用是为了活化硼酸,使其更好地发生转金属化.该案例成功开发了一种通过钯催化的未活化二氟烷基卤化物与烷基硼烷的羰基交叉偶联反应来获得烷基二氟烷基酮的有效方法.该反应克服了未活化二氟烷基卤化物的加氢脱卤和β-氢消除的副产物的产生,代表了未活化二氟烷基卤化物催化羰基化的第一个例子.二氟烷基碘代物与二氟烷基溴代物在此条件下均可很好地完成该反应,证明了该方法的普遍性.该反应的优点是合成简单、效率高、底物范围广和官能团耐受性高.因此,这种催化羰基化过程为药物化学中的应用提供了一条简便的途径,所得的烷基二氟烷基酮也可以进一步官能化修饰,从而实现二氟烷基化化合物的多样化合成.
2.1.2 Ni催化硼酸类碳亲核试剂参与的羰基化反应
2019年,Zhao等[52]首次实现了Ni催化芳基硼酸和溴代二氟烷基衍生物的羰基化反应(图42).该反应在温和反应条件下进行,充分体现了在Ni催化下的高选择性以及官能团耐受性好的特点.
羰基化反应可能的机理(图43)为:首先由[NiⅡ(Ln)X2]物种92与芳基硼酸的转金属化生成芳基鎳(Ⅱ)络合物93,然后CO迁移插入到络合物93中生成芳酰基镍(Ⅱ)络合物94.络合物94与二氟溴化物单电子转移生成二氟烷基自由基和芳酰基镍(Ⅲ)络合物95,接着二氟烷基自由基被芳酰基镍(Ⅱ)络合物94捕获,得到关键中间体96,之后还原消除生成二氟烷基酮化合物和镍(Ⅰ)配合物97,最后镍(Ⅰ)配合物97再与物种95通过歧化反应再生镍(Ⅱ)催化剂92和酰基镍(Ⅱ)络合物94来参与循环.
2019年,Zhao等[52]开发了镍催化芳基硼酸和溴二氟烷基化合物的羰基化反应,该羰基化反应可以在标准大气压的CO气氛及温和反应条件下顺利进行,提供了一种具有成本效益且直接获得二氟烷基酮的方法.值得注意的是,许多由此产生的含有C—C不饱和键的二氟烷基酮在此之前是未知的.
同年,Cheng等[53]又报道了在标准大气压的CO条件下,镍催化仲烷基卤化物与芳基硼酸的羰基化反应(图44).该反应体现了较高的官能团耐受性和广泛的底物范围,包括三氟甲基化、二氟甲基化和二氟乙酰化仲烷基碘化物和仲苄基溴化物均适用该催化体系,提供了一种获取烷基酮的新方式,具有α-三氟甲基酮的药物分子可以通过该方法有效合成.
2021年,Cheng等[54]再次使用Ni催化芳基硼酸与溴二氟烯烃在一个大气压的CO气氛下的交叉偶联羰基化反应(图45),得到了一系列高区域选择性的羰基化偕二氟烯烃产物100.该反应在温和条件下进行,具有高反应性和良好的官能团耐受性,合成的偕二氟烯烃还可作为底物进一步拓展.
2023年,Li等[55]报道了一种通过Ni催化1,5-氢原子转移策略的羰基化反应(图46),高效实现了远程C(sp3)-H活化羰基化,从而得到了一系列具有高度官能化的α-取代酮衍生物.此外,该反应可以在常压和氧化还原中性条件下顺利进行,并表现出良好的官能团相容性和优异的位点选择性.经过机理验证实验,提出了一种可能的反应途径(图46):首先,芳基硼酸在碱作用下与NiⅠ活性物种发生配位转金属化得到芳基NiⅠ物种103,随后与底物N-F酰胺101发生单电子转移得到酰胺基自由基107和NiⅡ物种104,之后酰胺基自由基107
经1,5氢原子转移得到苄基自由基108,NiⅡ物种104与CO发生配位迁移插入得到芳甲酰基NiⅡ物种105,苄基自由基108加成到芳甲酰基NiⅡ物种105上得到NiⅢ106物种,最后还原消除得到目标产物102并再生NiⅠ活性物种参与下一循环.
2020年,Zhou等[56]首次报道了标准大气压CO环境下,镍催化的四组分双官能团化羰基化反应(图47).使用各种芳基硼酸、烯烃和烷基亲电子试剂(包括二氟烷基溴化物和溴乙酸衍生物)合成了一系列复杂的羰基化合物,产生了高活性的氟化氨基酸和寡肽类衍生物.这种镍催化的羰基化串联过程克服了优先形成不活泼的Ni(CO)4物种的问题,为利用CO气体进行镍催化的羰基化反应开辟了一条新途径,其反应途径如图47所示:NiⅡ物种111与芳基硼酸配位转金属化得到中间体112,随后的CO配位插入得到物种113,其与溴二氟烷基试剂之间单电子转移产生相应的二氟烷基自由基和NiⅢ物种114.二氟烷基自由基与烯烃加成生成的自由基中间体115,再被NiⅡ物种113捕获得到中间体116,经还原消除得到目标化合物110和NiⅠ物种117,随后NiⅠ物种117与NiⅢ物种114通过歧化反应得到二次循环的NiⅡ中间体111参与下一循环.
2023年,Rao等[57]开发了一种镍催化溴二氟乙酸乙酯、芳基硼酸和缺电子烯烃(丙烯腈、 丙烯酸乙酯)的四组分二氟烷基羰基化反应(图48).该催化体系与CO气体高度相容,在Ar或CO气氛下均可生成相应的目标化合物,有效扩展了氟烷基化和含氟羰基化的合成途径,克服了先前缺电子烯烃参与多组分碳氟烷基化的局限性,具有合成简便、官能团耐受性高、原料简单易得等优点,可以方便地构建有价值的二氟烷基羰基化衍生物.
目前四组分羰基化反应主要集中于各类烯烃或炔烃的1,2-双官能团化反应,然而对于1,3-烯炔的1,4-烷基羰基化反应探索鲜有报道.2024年,Shan等[58]首次报道了在1 atm CO气氛下,通过Ni催化1,3-烯炔的1,4-烷基羰基化反应(图49).其中,对于1,3-烯炔的底物结构必须含有CF3,并以良好的收率和区域选择性提供了四取代的CF3-联烯酮化合物.该方案也是Ni催化在C(sp2)-C(sp2)上实现羰基化的第一个例子,而且有着条件温和、底物范围廣、官能团兼容性好等特点.
2.2 有机锌试剂碳亲核试剂
2023年,Weng等[59]报道了在标准大气压的
CO温和条件下,镍催化剂与有机锌试剂通过转金
属化羰基化生成酰基碳亲核试剂,再与不饱和烃通
过配位加成在原位生成酰胺、氧杂双环烯烃和α,β-不饱和酮的有机锌中间体,实现了高选择性的三组分酰基化反应(图50).所获得的有机锌中间体可以被各种功能化亲电试剂捕获,或进行分子内Truce-Smiles重排或羟醛缩合.该方法还能用于蒽环类抗生素柔红霉素的有效合成,以及制备具有聚集诱导发光活性的功能化1,3-二烯酮衍生物.通用的反应机理(图50)为:首先NiⅡ(L)X2与烷基锌试剂进行转金属化生成烷基NiⅡ(L)X物种134,随后CO通过配位迁移插入形成酰基NiⅡ(L)X物种135,然后酰基NiⅡ(L)X物种135再与不饱和键配位加成得到加成产物136,136再与烷基锌试剂发生转金属化得到新的烷基锌试剂137,最后再与亲电试剂发生Negishi偶联,或进行分子内Truce-Smiles重排或羟醛缩合的方式得到目标产物138.
有机金属试剂与CO气体结合可以作为酰基亲核试剂实现酰基化反应,有效拓展了烯烃碳环化反应的视野.2023年,Wang等[60]开发了镍催化的苯乙烯衍生物139与烷基锌试剂在常压CO条件下的酰基化、环化反应(图51).值得注意的是,在酰基镍物种向邻异氰基肉桂酸酯或邻位异氰基苯乙烯的双键进行配位加成中,会发生区域选择性的逆转,最终生成不同官能化的环化结构,从而为构建医学相关的小分子提供了一种有效的合成途径.
2024年,Zhang等[61]报道了在一个大气压的CO气氛中,通过镍催化实现的二级碘代物与有机碘化锌试剂的交叉羰基化偶联反应(图52).新型三齿配体是该反应顺利转化的关键,有效克服了Negishi偶联和β-H消除等副产物的产生.
3 过渡金属催化碳亲电试剂参与的羰基化反应
2015年,Makarov等[62]首次报道了钯催化的乙酰乙酸的二烯酸衍生物和芳基或乙烯基碘化物的羰基化反应(图53),生成γ-位酰基取代的3,5-二羰基酸和2,2,6-三甲基-4H-1,3-二噁英-4-酮.该反应在室温下通过双室反应器发生,使该反应高效且具有γ-选择性.值得一提的是,当噁酮的γ-位有甲基代基时,苯甲酰基和烯丙酰基取代的目标产物也能以中等的产率生成,同时苯基取代基位于二噁酮的α-位时,能够以优异的产率获得苯甲酰基取代的酮类化合物,而烯丙酰基取代的目标产物产率适中.同时,该方法可以应用于一系列HMGCoA还原酶抑制剂和他汀类药物的合成以及13C的同位素标记,为药物分子的合成奠定了基础.
茚满酮的骨架普遍存在于天然产物中,是一种价值较高的合成子和药效基团.2019年,Cai等[63]报道了一种钯催化的芳基三氟甲磺酸酯或芳基溴化物在1 atm CO下的C(sp3)-H羰基化反应(图54).该反应不需要导向基团或氧化剂,钯催化剂与NHC配体的催化体系有效促进了五元环钯中间体的形成.机理研究表明,CO插入到五元环钯物种可能是该羰基化反应的关键步骤.
2021年,Zhao等[64]报道了钯催化下含有弱酸性苄基C(sp3)-H键的亲核试剂-氮杂芳基甲基胺和溴苯的芳基羰基化反应(图55),使用Pd/NiXANTPHOS为催化体系,通过苄基C-H键的原位去质子化过程,以广泛的底物范围、良好的官能团耐受性以及中等至较好的产率一锅法偶联制备α-氨基芳基-氮杂芳基甲基酮化合物.
2023年,Hu等[65]报道了一种以LiHMDS(双(三甲基硅基)胺基锂)为碱的Pd催化体系来实现苄基C(sp3)-H与芳基溴化物的羰基化反应(图56).这种新的催化剂体系能够偶联具有高pKa值(在DMSO中接近pKa 32值)的亲核试剂.低负载量催化剂(1 mol %)和常压反应(1 atm CO)是该反应的一大亮点,增加了其工业应用的潜力.
4 过渡金属催化的还原偶联羰基化反应
2018年,Peng等[66]报道了钯催化芳基碘化物与烷基溴化物的还原性羰基化反应(图57),以甲酸为羰基源,利用易得的烷基和芳基卤化物以中等到良好的收率合成了一系列烷基芳基酮化合物.其中烷基溴化物在Mg和ZnCl2作用下可以更好的生成有机锌试剂,更容易发生Negishi偶联型羰基化反应.其中伯和仲烷基溴化物/碘化物都可实现两种亲电试剂的羰基化偶联反应.该方法还可用于复杂天然产物和多功能分子的后期修饰功能化.
2023年,Zhang等[67]又实现了一种在低负载量钯催化剂的温和条件下,芳基噻蒽盐与苄基氯在Zn粉还原下进行芳烃位点选择性Negishi型羰基化反应(图58),反应以良好产率得到了所需的1,2-二芳基乙基酮,还可以实现复杂分子的后期修饰,这种方法在Negishi型羰基化反应中极具挑战性.可能的反应机理为:首先Pd(0)与芳基噻蒽盐88发生氧化加成得到PdⅡ中间体152,随后CO配位迁移插入得到酰基PdⅡ中间体153,苄基氯与Zn粉的作用下生成苄基氯化锌,并与酰基PdⅡ中间体154发生转金属化、还原消除得到最终产物.
镍催化常被用于还原偶联反应中.2018年,Peng等[68]报道了一种镍催化条件下,以六羰基合钼为羰基源的还原偶联羰基化反应(图59),由芳基碘化物以中等到极好的收率合成了各种取代的对称二芳基酮化合物,其中镍和钼的协同作用是这种高效转变成功的原因.当使用两种不同的芳基碘化物进行反应时,通过改变两种底物的比例,均不能以较高的产率得到交叉羰基化产物.
2020年,Geng等[69]报道了镍催化还原偶联羰基化合成二氢苯并呋喃衍生物(图60).以Mo(CO)6作为CO源、金属锰作为还原剂,实现了烷基卤化物与芳基邻碘烯丙醚类化物的还原偶联型羰基化反应,以中等到良好的收率得到所需的产物.他们提出了一种可能的反应机理(图61):首先Ni(acac)2在金属Mn和配体的作用下生成Ni0L物
种,然后Ni0L络合物与芳基邻碘烯丙醚155氧化
加成形成芳基NiⅡ络合物157,157经过分子内环化、加成形成烷基NiⅡ络合物158,158与金属Mn还原成烷基NiⅠ络合物159,接着烷基碘与络合物159发生氧化加成,得到烷基NiⅢ中间体160,羰基钼释放一氧化碳并迁移插入到烷基NiⅢ中间体160得到中间体161,最后还原消除得到最终的目标产物和NiⅠL物种.NiⅠL中间体被Mn进一步还原为Ni0L物种,参与下一个催化循环.
2019年,Shi等[70]首次报道了一种用于合成二烷基酮的镍催化三组分还原偶联羰基化方法(图62).从两种烷基卤化物出发,用氯甲酸乙酯作为安全的羰基源获得范围广泛的不对称或对称的二烷基酮化合物.该羰基化反应在温和的反应条件下发生,具有广泛底物范围和高官能团耐受性,为现有的酮类化合物的合成方法提供了有价值的补充.
同年,Xu等[71]使用氯甲酸异丁酯作为新型的羰基源,实现了Ni催化Heck型环化策略的还原偶联羰基化反
应(图63),以温和条件制备了一系列含有手性季碳中心的吲哚酮类衍生物162.
2022年,Chen等[72]报道了镍催化卤代烷、卤代芳烃和氯甲酸乙酯的三组分还原偶联羰基化反应(图64).氯甲酸乙酯作为一种安全易得的CO源被用于该多组分反应方案中,为芳基烷基酮的合成提供了一种高效实用的替代方案.该反应具有广泛的底物范围和良好的官能团相容性.通过DFT机理研究突出了交叉亲电偶联羰基化反应的复杂性,并加深了对芳基卤化物、氯甲酸酯和烷基卤化物的三个连续氧化加成顺序的深入了解.
根据DFT研究,提出了该反应的可能机理(图64):NiⅡ预催化剂被锌还原为Ni0,然后与NiⅡ发生歧化反应生成NiI-Ⅰ中间体163,随后芳基碘化物与NiI中间体163发生氧化加成,歧化得到Ar-NiⅡ-Ⅰ中间体164.芳基碘也可以与Ni0直接进行氧化加成得到164,这是在初始催化循环中生成中间体164的另一种方法.生成的中间体164进一步被锌还原生成Ar-NiⅠ中间体165.氯甲酸乙酯与Ar-NiⅠ中间体165发生氧化加成得到NiⅢ中间体166,其被锌还原为NiⅡ中间体167.乙氧基发生迁移形成新的NiⅡ中间体168,进一步被锌还原以提供Ar-NiⅠ-CO中间体169.中间体169与烷基溴化物发生单电子转移得到NiⅡ中间体170和烷基自由基.CO迁移插入形成关键的ArCO-NiⅡ-Br中间体171,烷基自由基被中间体171捕获形成ArCO-NiⅢ-alkyl中间体172.最后经过还原消除得到最终产物和NiⅠ物种163参与下一催化循环.
酰基糖苷是天然產物和药用相关物质的通用中间体,通常这类化合物的合成依赖于常规交叉偶联策略,在过渡金属或双催化条件下由糖基前体与酰基供体(预合成或原位生成)偶联形成C-C键的方式合成.2022年,Jiang等[73]报道了一种三组分Ni催化的还原偶联羰基化反应机制(图65),使用氯甲酸异丁酯作为羰基源,促进了糖基卤化物和有机碘化物还原偶联羰基化反应.该方法具有良好的选择性以及广泛的底物适用性,并且以高非对映选择性获得了目标产物.为糖类羰基化化学开辟了新途径.
NiH催化的多组分烯烃加氢还原官能化是一种有吸引力但探索不足的快速增加分子复杂性的方法.2021年,Chen等[74]开发了一种镍催化的多组分对映选择性羰基化偶联反应(图66).该体系使用仲苄基氯和α-氨基酸的NHP酯作为亲电试剂,氯甲酸酯作为羰基源,通过硅氢试剂的加氢官能化,使多种未活化的烯烃直接实现氢化、羰基化反应.该反应在双噁唑啉为手性配体的条件下,产生了一系列带有α-立体中心的不对称烷基酮衍生物.
电催化还原性亲电交叉偶联是一种强大、高效的构建C—C键的新方法,这种方法大多数报道以两种亲电试剂或加入第三组分(不饱和烃)实现双官能团化的还原偶联.2023年,Xie等[75]首次报道了在电催化条件下,烷基卤化物、芳基碘化物和氯甲酸丙酯的三组分顺序偶联反应(图67).该反应无需添加剂和活化剂,同时还有效避免了化学计量金属还原剂及有毒的CO气体或金属羰基络合物的使用.
草酰氯作为一种廉价的市售化学品,是化学转化中用途最广泛的有机试剂之一.2023年,Wang等[76]开发了以草酰氯为CO源,使用烷基卤化物和芳基碘化物两种不同的亲电试剂还原偶联羰基化反应(图68),有效获得了广泛的烷基芳基酮衍生物,该方案也可以用于天然产物和药物分子的衍生化.
5 结束语
尽管过渡金属催化羰基化反应取得了重大进展,但多组分羰基化反应的发展仍具有进一步探索的空间.高化学选择性、对映选择性,以及多样性的羰基化策略都彰显了过渡金属催化羰基化反应的价值.因此,挖掘出更多形式的多组分羰基化反应,开发效率更高、条件更温和的过渡金属催化羰基化反应的方案势在必行.截至目前,羰基化反应中主要采用Pd催化体系,这主要是因为Pd催化羰基化反应的活性高、体系兼容性较强.此外,文中还总结了近年来相对廉价金属Cu,Co,Mn,Fe和Ni催化羰基化转化的主要研究成果.展望未来,探索开发更加温和的反应体系,实现安全、绿色的多组分的羰基化反应在有机合成中变得越来越有价值.
参考文献:
[1] MA K,MARTIN B S,YIN X,et al.Natural product syntheses via carbonylative cyclizations[J].Nat Prod Rep,2019,36(1):174.
[2] LI W,JIANG D,WANG C,et al.Recent advances in base-metal-catalyzed carbonylation of unactivated alkyl electrophiles[J].Chin J Chem,2023,41(23):3419.
[3] SHEN C,WU X F.Palladium-catalyzed carbonylative multicomponent reactions[J].Chem Eur J,2017,23(13):2973.
[4] TIAN Q,YIN X,SUN R,et al.The lower the better:Efficient carbonylative reactions under atmospheric pressure of carbon monoxide[J].Coord Chem Rev,2023,475:214900.
[5] WU X F,FANG X,WU L,et al.Transition-metal-catalyzed carbonylation reactions of olefins and alkynes:A personal account[J].Acc Chem Res,2014,47(4):1041.
[6] PENG J B,WU F P,WU X F.First-row transition-metal-catalyzed carbonylative transformations of carbon electrophiles[J].Chem Rev,2019,119(4):2090.
[7] PENG J B,GENG H Q,WU X F.The chemistry of CO:Carbonylation[J].Chem,2019,5(3):526.
[8] SCHOENBERG A,HECK R F.Palladium-catalyzed amidation of aryl,heterocyclic,and vinylic halides[J].J Org Chem,1974,39(23):3327.
[9] 王進贤.羰基锰催化羰化炔烃合成不饱和内酯[J].西北师范大学学报(自然科学版),1994,30(3):114.
[10] 王进贤,张玉梅,胡雨来.羰基钴催化羰化炔烃合成γ-酮酸[J].西北师范大学学报(自然科学版),1998,34(1):91.
[11] WANG L C,CHEN B,ZHANG Y,et al.Nickel-catalyzed four-component carbonylation of ethers and olefins:Direct access to γ-oxy esters and amides[J].Angew Chem Int Ed,2022,61(33):e202207970.
[12] DING Y,HUANG R,ZHANG W,et al.Nickel-catalyzed oxidative carbonylation of alkylarenes to arylacetic acids[J].Org Lett,2022,24(43):7972.
[13] ZHOU F,DAI H,TANG S,et al.Synthesis of polyethylenes with in-chain isolated carbonyls from CO2 and ethylene via a tandem photoreduction/polymerization protocol[J].CCS Chem,2023,doi:10.31635/ccschen.023.202303275.
[14] LI Y,HU Y,WU X F.Non-noble metal-catalysed carbonylative transformations[J].Chem Soc Rev,2018,47(1):172.
[15] XU J X,WANG L C,WU X F.Non-noble metal-catalyzed carbonylative multi-component reactions[J].Chem Asian J,2022,17(22):e202200928.
[16] ZHANG S,NEUMANN H,BELLER M.Pd-catalyzed carbonylation of vinyl triflates to afford α,β-unsaturated aldehydes,esters,and amides under mild conditions[J].Org Lett,2019,21(10):3528.
[17] WANG P,YANG J,SUN K,et al.A general synthesis of aromatic amides via palladium-catalyzed direct aminocarbonylation of aryl chlorides[J].Org Chem Front,2022,9(9):2491.
[18] BAO Z P,WU X F.Palladium-catalyzed direct carbonylation of bromoacetonitrile to synthesize 2-cyano-n-acetamide and 2-cyanoacetate compounds[J].Angew Chem Int Ed,2023,62(19):e202301671.
[19] WANG H,WU X F.Palladium-catalyzed carbonylative dearomatization of indoles[J].Org Lett,2019,21(13):5264.
[20] CHEN M,WANG X,YANG P,et al.Palladium-catalyzed enantioselective heck carbonylation with a monodentate phosphoramidite ligand:Asymmetric synthesis of(+)-physostigmine,(+)-physovenine,and(+)-folicanthine[J].Angew Chem Int Ed,2020,59(29):12199.
[21] YUAN Z,ZENG Y,FENG Z,et al.Constructing chiral bicyclo[3.2.1] octanes via palladium-catalyzed asymmetric tandem heck/carbonylation desymmetrization of cyclopentenes[J].Nat Commun,2020,11(1):2544.
[22] HU H,YU T,CHENG S,et al.Palladium-catalyzed tandem heck/carbonylation/aminocarbonylation en route to chiral heterocyclic α-ketoamides[J].Org Chem Front,2022,9(4):939.
[23] LI Q,ZHANG Y,ZENG Y,et al.Palladium-catalyzed asymmetric dearomative carbonylation of indoles[J].Org Lett,2022,24(16):3033.
[24] CHEN L,SHI C,LI W,et al.Palladium-catalyzed asymmetric C-C bond activation/carbonylation of cyclobutanones[J].Org Lett,2022,24(49):9157.
[25] LI L,LIU X L,LIANG J Y,et al.Palladium catalyzed dicarbonylation of α-iodo-substituted alkylidenecyclopropanes:Synthesis of carbamoyl substituted indenones[J].Org Lett,2022,24(30):5624.
[26] CˇARNY T,ROCABOY R,CLEMENCEAU A,et al.Synthesis of amides and esters by palladium(0)-catalyzed carbonylative C(sp3)-H activation[J].Angew Chem Int Ed,2020,59(43):18980.
[27] LI M,LI S X,CHEN D P,et al.Regioselective C-H active carbonylation via 1,4-palladium migration[J].Org Lett,2023,25(16):2761.
[28] WANG Q,HE Y T,ZHAO J H,et al.Palladium-catalyzed regioselective difluoroalkylation and carbonylation of alkynes[J].Org Lett,2016,18(11):2664.
[29] ZHANG Y,GENG H Q,WU X F.Palladium-catalyzed carbonylative four-component synthesis of β-perfluoroalkyl amides[J].Chem Eur J,2021,27(70):17682.
[30] CHENG S,LUO Y,YU T,et al.Palladium-catalyzed four-component cascade imidoyl-carbamoylation of unactivated alkenes[J].ACS Catal,2022,12(2):837.
[31] AI H J,WANG H,LI C L,et al.Rhodium-catalyzed carbonylative coupling of alkyl halides with phenols under low CO pressure[J].ACS Catal,2020,10(9):5147.
[32] AI H J,RABEAH J,BRCKNER A,et al.Rhodium-catalyzed carbonylative coupling of alkyl halides with thiols:A radical process faster than easier nucleophilic substitution[J].Chem Commun,2021,57(12):1466.
[33] WANG P,WANG Y,NEUMANN H,et al.Rh-catalyzed alkoxycarbonylation of unactivated alkyl chlorides[J].Chem Sci,2022,13(45):13459.
[34] CHEN Y,SU L,GONG H.Copper-catalyzed and indium-mediated methoxycarbonylation of unactivated alkyl iodides with balloon CO[J].Org Lett,2019,21(12):4689.
[35] LU B,CHENG Y,CHEN L Y,et al.Photoinduced copper-catalyzed radical aminocarbonylation of cycloketone oxime esters[J].ACS Catal,2019,9(9):8159.
[36] YIN Z,ZHANG Y,ZHANG S,et al.Copper-catalyzed intra- and intermolecular carbonylative transformation of remote C(sp3)H bonds in n-fluoro-sulfonamides[J].J Catal,2019:377507.
[37] ZHANG Y,YIN Z,WU X F.Copper-catalyzed carbonylative synthesis of β-homoprolines from n-fluoro-sulfonamides[J].Org Lett,2020,22(5):1889.
[38] ZHANG Y,AI H J,WU X F.Copper-catalyzed carbonylative synthesis of pyrrolidine-containing amides from γ,δ-unsaturated aromatic oxime esters[J].Org Chem Front,2020,7(19):2986.
[39] ZHANG Y,BAO Z P,KUAI C S,et al.Copper-catalyzed visible-light-induced ring-opening carbonylation of sulfonium salts[J].J Catal,2023,426:1.
[40] ZHAO F,AI H J,WU X F.Copper-catalyzed substrate-controlled carbonylative synthesis of α-keto amides and amides from alkyl halides[J].Angew Chem Int Ed,2022,61(17):e202200062.
[41] WU F P,YUAN Y,WU X F.Copper-catalyzed 1,2-trifluoromethylation carbonylation of unactivated alkenes:Efficient access to β-trifluoromethylated aliphatic carboxylic acid derivatives[J].Angew Chem Int Ed,2021,60(49):25787.
[42] ZHANG Y,YUAN Y,GENG H Q,et al.Visible light-induced perfluoroalkylative carbonylation of unactivated alkenes[J].J Catal,2022,413:214.
[43] ZHANG Y,TENG B H,WU X F.Copper-catalyzed trichloromethylative carbonylation of ethylene[J].Chem Sci,2024,15(4):1418.
[44] AI H J,YUAN Y,WU X F.Ruthenium pincer complex-catalyzed heterocycle compatible alkoxycarbonylation of alkyl iodides:Substrate keeps the catalyst active[J].Chem Sci,2022,13(8):2481.
[45] AI H J,LEIDECKER B N,DAM P,et al.Iron-catalyzed alkoxycarbonylation of alkyl bromides via a two-electron transfer process[J].Angew Chem Int Ed,2022,61(43):e202211939.
[46] AI H J,ZHAO F,WU X F.SET or TET?Iron-catalyzed aminocarbonylation of unactivated alkyl halides with amines,amides,and indoles via a substrate dependent mechanism[J].Chin J Catal,2023:47121.
[47] AI H J,GENG H Q,GU X W,et al.Manganese-catalyzed alkoxycarbonylation of alkyl chlorides[J].ACS Catal,2023,13(2):1310.
[48] ZHAO H Y,FENG Z,LUO Z,et al.Carbonylation of difluoroalkyl bromides catalyzed by palladium[J].Angew Chem Int Ed,2016,55(35):10401.
[49] ZHANG S,NEUMANN H,BELLER M.Pd-catalyzed synthesis of α,β-unsaturated ketones by carbonylation of vinyl triflates and nonaflates[J].Chem Commun,2019,55(42):5938.
[50] ZHANG J,WU X F.Palladium-catalyzed carbonylative synthesis of diaryl ketones from arenes and arylboronic acids through C(sp2)-H thianthrenation[J].Org Lett,2023,25(12):2162.
[51] ZHAO H Y,ZHOU M,ZHANG X.Palladium-catalyzed carbonylative cross-coupling of difluoroalkyl halides with alkylboranes under 1 atm of CO[J].Org Lett,2021,23(23):9106.
[52] ZHAO H Y,GAO X,ZHANG S,et al.Nickel-catalyzed carbonylation of difluoroalkyl bromides with arylboronic acids[J].Org Lett,2019,21(4):1031-1036.
[53] CHENG R,ZHAO H Y,ZHANG S,et al.Nickel-catalyzed carbonylation of secondary trifluoromethylated,difluoromethylated,and nonfluorinated aliphatic electrophiles with arylboronic acids under 1 atm of CO[J].ACS Catal,2020,10(1):36.
[54] CHENG R,SANG Y,GAO X,et al.Highly γ-selective arylation and carbonylative arylation of 3-bromo-3,3-difluoropropene via nickel catalysis[J].Angew Chem Int Ed,2021,60(22):12386.
[55] LI M,GAO F,MIAO D-Y,et al.Redox neutral radical-relay nickel-catalyzed remote carbonylation[J].Org Lett,2023,25(13):2306.
[56] ZHOU M,ZHAO H Y,ZHANG S,et al.Nickel-catalyzed four-component carbocarbonylation of alkenes under 1 atm of CO[J].J Am Chem Soc,2020,142(42):18191.
[57] RAO N,LI Y Z,LUO Y C,et al.Nickel-catalyzed multicomponent carbodifluoroalkylation of electron-deficient alkenes[J].ACS Catal,2023,13(7):4111.
[58] SHAN Q C,ZHAO Y,WANG S T,et al.Nickel-catalyzed modular four-component 1,4-alkylcarbonylation of 1,3-enynes to tetra-substituted CF3-allenyl ketones[J].ACS Catal,2024,14(4):2144.
[59] WENG Y,ZHANG Y,TURLIK A,et al.Nickel-catalysed regio- and stereoselective acylzincation of unsaturated hydrocarbons with organozincs and CO[J].Nat Synth,2023,2(3):261.
[60] WANG C,LIU N,WU X,et al.Nickel-catalyzed regiodivergent acylzincation of styrenes with organozincs and CO[J].Chin J Chem,2024,42(6):599.
[61] ZHANG Y,CAO Q,XI Y,et al.Nickel-catalyzed carbonylative negishi cross-coupling of unactivated secondary alkyl electrophiles with 1 atm CO gas[J].J Am Chem Soc,2024,146(12):7971.
[62] MAKAROV I S,KUWAHARA T,JUSSEAU X,et al.Palladium-catalyzed carbonylative couplings of vinylogous enolates:application to statin structures[J].J Am Chem Soc,2015,137(44):14043.
[63] CAI S L,LI Y,YANG C,et al.NHC ligand-enabled,palladium-catalyzed non-directed C(sp3)-H carbonylation to access indanone cores[J].ACS Catal,2019,9(11):10299.
[64] ZHAO H,HU B,XU L,et al.Palladium-catalyzed benzylic C(sp3)-H carbonylative arylation of azaarylmethyl amines with aryl bromides[J].Chem Sci,2021,12(32):10862.
[65] HU B,ZHAO H,WU Y,et al.Palladium-catalyzed benzylic C(sp3)-H carbonylative arylation with aryl bromides[J].Angew Chem Int Ed,2023,62(23):e202300073.
[66] PENG J B,CHEN B,QI X,et al.Palladium-catalyzed carbonylative coupling of aryl iodides with alkyl bromides:efficient synthesis of alkyl aryl ketones[J].Adv Synth Catal,2018,360(21):4153.
[67] ZHANG J,WANG L C,BAO Z P,et al.Site-selective carbonylation of arenes via C(sp2)-H thianthrenation:direct access to 1,2-diarylethanones[J].Chem Sci,2023,14(28):7637.
[68] PENG J B,WU F P,LI D,et al.Nickel-catalyzed molybdenum-promoted carbonylative synthesis of benzophenones[J].J Org Chem,2018,83(12):6788.
[69] GENG H Q,WANG W,WU X F.Nickel-catalyzed carbonylative synthesis of dihydrobenzofurans[J].Catal Commun,2021:148106170.
[70] SHI R,HU X.From alkyl halides to ketones:Nickel-catalyzed reductive carbonylation utilizing ethyl chloroformate as the carbonyl source[J].Angew Chem Int Ed,2019,58(22):7454.
[71] XU S,WANG K,KONG W.Ni-catalyzed reductive arylacylation of alkenes toward carbonyl-containing oxindoles[J].Org Lett,2019,21(18):7498.
[72] CHEN H,YUE H,ZHU C,et al.Reactivity in nickel-catalyzed multi-component sequential reductive cross-coupling reactions[J].Angew Chem Int Ed,2022,61(33):e202204144.
[73] JIANG Y,YANG K,WEI Y,et al.Catalytic multicomponent synthesis of C-acyl glycosides by consecutive cross-electrophile couplings[J].Angew Chem Int Ed,2022,61(46):e202211043.
[74] CHEN J,ZHU S.Nickel-catalyzed multicomponent coupling:synthesis of α-chiral ketones by reductive hydrocarbonylation of alkenes[J].J Am Chem Soc,2021,143(35):14089.
[75] XIE S,YIN Y,WANG Y,et al.Current-controlled nickel-catalyzed multi-electrophile electroreductive cross-coupling[J].Green Chem,2023,25(4):1522.
[76] WANG J,YIN Y,HE X,et al.Nickel-catalyzed highly selective reductive carbonylation using oxalyl chloride as the carbonyl source[J].ACS Catal,2023,13(12):8161.
(責任编辑 陆泉芳)
猜你喜欢
江苏安全生产(2024年2期)2024-03-13
小星星·阅读100分(低年级)(2023年9期)2023-10-28
山东青年报·教育周刊教师版上半月(2022年52期)2022-03-10
原子与分子物理学报(2021年2期)2021-03-29
国际呼吸杂志(2019年8期)2019-04-29
消费电子(2016年10期)2016-11-10
压缩机技术(2014年3期)2014-02-28
河南科技(2014年12期)2014-02-27
河北医科大学学报(2011年8期)2011-03-25
祝您健康(1989年1期)1989-12-30
