
氯沙坦的合成*
2010-11-26 03:23闫起强郭拥政谢苏豪王文峰
合成化学 2010年1期
闫起强, 郭拥政, 谢苏豪, 祁 伟, 杨 琰, 王文峰
(1. 北京赛科药业有限公司 研发部,北京 100021; 2. 浙江新赛科药业有限公司,浙江 上虞 312369)
氯沙坦(1)的钾盐是第一个沙坦类抗高血压药,由美国杜邦和默克联合公司开发,1994年11月首先在瑞典获准上市,1995年4月14日获FDA批准,并相继在许多国家上市[1]。1的钾盐是第一个上市的非肽类血管紧张素Ⅱ受体拮抗剂,药理和临床试验表明其具有作用广泛、降压作用显著、服药方便、对肾功能影响小等优点[2]。
1的合成方法[1,3~7]主要有三类: (1) 2-丁基-4-氯-5-羟甲基咪唑(2)与对溴苄溴反应生成[3-(4-溴-1-苄基)-2-丁基-5-氯-3H-咪唑-4-基]甲醛;再与2-(2H-四氮唑-5-基)苯硼酸反应生成三苯基氯沙坦(4),后者再还原、脱保护基得1。此路线收率低(总收率35%),反应步骤多,反应条件苛刻,且需使用钯等昂贵催化剂等都限制了其在工业上的应用。(2)2与2′-氰基-4-溴甲基联苯进行氮烷基化还原生成2-丁基-4-氯-5-羟甲基-1-[(2′-氰基)-联苯-4-甲基] 咪唑;再与叠氮化钠在高温下形成四氮唑得1。此路线反应步骤少,收率高(总收率76%),但环合反应使用的原料叠氮化钠是剧毒化学品,后处理复杂,且对环境破坏大。(3)2与5-(4-溴甲基联苯-2-基)-1-三苯甲基-1H-四唑(3)进行氮烷基化反应生成4,然后再还原脱保护基得1, 此路线反应条件温和,操作简单,但收率较低(总收率45%~50%)。
本文综合文献方法合成1(Scheme 1),2与3反应生成4;4先脱保护基生成2-丁基-4-氯-5-甲酰基-1-{[2′-(1-H-四唑-5-基)-联苯-4]-甲基}咪唑5); 5经还原得1,总收率64%,其结构经1H NMR表征。


Scheme1
1 实验部分
1.1 仪器与试剂
MERCURY plus-400型核磁共振仪(CDCl3为溶剂,TMS为内标);LC-10AT型高效液相色谱仪。
2,工业级,盐城市药物化工厂;3,工业级,浙江天宇化工有限公司;石油醚,60 ℃~90 ℃;其余所用试剂均为分析纯。
1.2 合成
(1) 4的合成
在三口瓶中加入2 33.5 g(179 mmol),3100 g(179 mmol), DMF 300 mL,碳酸钾47 g(340 mmol),于30 ℃反应24 h。将反应液倒入水(600 mL)中,抽滤,滤饼用水(100 mL)洗涤,于60 ℃烘48 h得白色固体4107 g,收率90%(粗品不需处理,直接进行下一步反应), m.p.130 ℃~133 ℃;1HNMRδ: 0.86( t,J=7.2 Hz, 3H, CH3), 1.24~1.34(m, 2H, CH2), 1.60~1.68(m, 2H, CH2), 2.52(t,J=7.6 Hz, 2H, CH2), 5.45(s, 2H, CH2), 6.83(d,J=8.0 Hz, 2H, CH), 6.91(d,J=8.0 Hz, 6H, CH), 7.11(d,J=8.0 Hz, 2H, CH), 7.23~7.50(m, 12H, CH), 7.95(d,J=8.0 Hz, 1H, CH), 9.74(s, 1H, CHO)。
(2) 5的合成
在反应瓶中加入482 g(123 mmol)的异丙醇(420 mL)溶液,搅拌下滴加3.4 mol·L-1盐酸40 mL(136 mmol, 10 min内),于室温反应24 h。用10 mol·L-1NaOH溶液调至pH 13,蒸干异丙醇后加入去离子水206 mL,分液,水层用甲苯(2×100 mL)洗涤后用3.4 mol·L-1盐酸调至pH 3.0;加入二氯甲烷200 mL,搅拌30 min,静置分液,有机层用无水硫酸镁干燥,减压蒸除溶剂,残余物经柱层析[洗脱剂:V(乙酸乙酯) ∶V(石油醚)=3 ∶1]分离得淡黄色固体542 g,收率80%, m.p.68 ℃~70 ℃;1HNMRδ: 0.84(t,J=7.2 Hz, 3H, CH3), 1.22~1.34(m, 2H, CH2), 1.56~1.64(m, 2H, CH2), 2.58(t,J=8.0 Hz, 2H, CH2), 5.48(s, 2H, CH2), 6.94(d,J=8 Hz, 2H, CH), 7.05(d,J=8.0 Hz, 2H, CH), 7.37(t,J=7.6 Hz, 1H, CH), 7.48(t,J=7.6 Hz, 1H, CH), 7.57(t,J=7.6 Hz, 1H, CH), 7.85(d,J=7.6 Hz, 1H, CH), 9.6(s, 1H, CHO)。
(3) 1的合成
在反应瓶中加入542 g(100 mmol)的异丙醇(200 mL)溶液,搅拌下缓缓加入硼氢化钾5.4 g(100 mmol),加毕,于30 ℃反应10 h。减压蒸干溶剂,加去离子水100 mL,用10 mol·L-1NaOH溶液调至pH 13,分液;水层用二氯甲烷(2×100 mL)洗涤后加入乙酸乙酯80 mL,于10 ℃用3.4 mol·L-1盐酸调至pH 3.0,搅拌1 h;抽滤,滤饼于60 ℃真空干燥30 h得白色固体1 38 g,收率89%,纯度99.2%(HPLC), m.p.182 ℃~183 ℃(Merck 索引183.5 ℃~ 184.5 ℃);1HNMRδ: 0.87(t,J=7.2 Hz, 3H, CH3), 1.24~1.36(m, 2H,CH2), 1.48~1.69(m, 2H, CH2), 2.39(t,J=7.2 Hz , 2H, CH2), 4.43(s, 2H, CH2), 5.16(s, 2H, CH2), 6.87(d,J=8.0 Hz, 2H, CH), 7.04(d,J=8.0 Hz, 2H, CH), 7.35~7.55(m, 3H, CH), 7.80~7.90(m, 1H, CH)。
2 结果与讨论
4的脱保护基反应(4→5)是整个合成路线的关键步骤,反应通常在甲醇[1,8],THF[3]以及丙酮[9]或者混合溶剂中[11]进行,常用酸盐酸或硫酸[8~11],鉴于THF沸点较低且价格较贵,丙酮在酸性条件下有自身缩合倾向[12],因此醇成为首选。我们在研究中发现,用甲醇作为溶剂会发生副反应,无法避免生成副产物,且随反应时间的增加或反应温度的提高副产物还会增加,从而影响1的品质。我们推测,副反应是1的四氮唑与甲醇在酸性条件下发生的氮甲基化反应,副产物为6或7(Chart 1)。
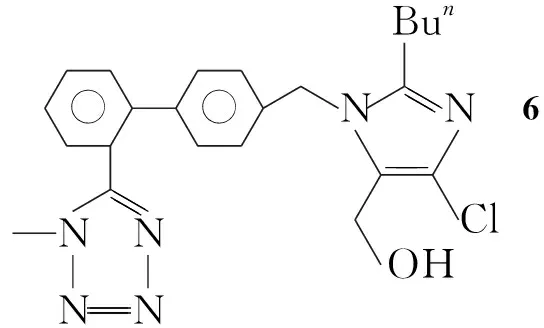

Chart1
为了证明这个推测,我们合成了6和7,并通过HPLC将6,7与Scheme 1的副产物在同一条件下进行了分析对比,确定副产物为7。因此我们选择空间位阻较大的异丙醇代替甲醇作溶剂,副产物很少,副反应得到了有效控制。这说明在4的脱保护基反应中,使用C3~C6空间位阻较大的仲醇或叔醇代替直链醇作溶剂是一个减少副反应,提高产品品质的好方法。
6:1H NMRδ: 0.89(t,J=7.2 Hz, 3H, CH3), 1.31~1.39(m, 2H, CH2), 1.61~1.68(m, 2H, CH2), 2.54(t,J=7.2 Hz, 2H, CH2), 3.28(s, 3H, CH3), 4.50(s, 2H, CH2), 5.20(s, 2H, CH2), 6.95(d,J=7.6 Hz, 2H, CH), 7.11(d,J=7.6 Hz, 2H, CH), 7.35~7.55(m, 3H, CH), 7.69(d,J=7.6 Hz, 1H, CH)。
7:1H NMRδ: 0.89(t,J=7.2 Hz, 3H, CH3), 1.31~1.36(m, 2H, CH2), 1.63~1.67(m, 2H, CH2), 2.57(t,J=7.2 Hz, 2H, CH2), 4.2(s, 3H, CH3), 4.50(s, 2H, CH2), 5.20(s, 2H, CH2), 6.95(d,J=8.0 Hz, 2H, CH), 7.15(d,J=8 Hz, 2H, CH), 7.35~7.55(m, 3H, CH), 7.81~7.83(d,J=8.0 Hz, 1H, CH)。
3 结论
以2-丁基-4-氯-5-羟甲基咪唑和5-(4-溴甲基联苯-2-基)-1-三苯甲基-1H-四唑为原料,通过氮烷基化、脱保护基和还原反应合成了氯沙坦,总收率64%。脱保护基反应宜选择C3~C6空间位阻较大的仲醇或叔醇代替直链醇作溶剂。
该路线具有原材料易得,反应条件温和,操作简单,环境友好,反应收率高的特点。
[1] Carrini D, Dunica J, Jonas V,etal. Angiotensin Ⅱ receptor blocking imidazoles[P].EP 0 253 310,1988.
[2] Loa L, Wagslaff J, Anlona. Losartan potassium: a review of its pharmacology, clinic efficacy and tolerability in the management of hepertansion[J].Drugs,1995,51(6):820-824.
[3] Young S Lo, Stenning D, Hocksesin, Del,etal. Tetrazophenylboronic acid intermediates for the synthesis of all receptor antagonists[P].US 5 130 439,1992.
[4] Allegrini P. A process for the preparation of angiotensin Ⅱ antagonistic compounds[P].EP 1 777 224,2007.
[5] Veera R, Udaya B, Rajendiran C,etal. A new process for the preparation of losartan[P].WO 2 007 020 654,2007.
[6] Veera R, Udaya B, Rajendiran C,etal. Process for the preparation of losartan[P].WO 2 007 026 375,2007.
[7] 王亚平,郑国君,蔡国华,等. 一种制备洛沙坦的方法[P].CN 1 915 990,2007.
[8] Khamar B, Modl I, Madhusudana R,etal. Process for the preparation of losartan potassium form Ⅰ[P].WO 2 005 023 758,2005.
[9] Radl S, Stach J, Klecan O,etal. A method of removing the triphenylmethane protecting group[P].WO 2 005 021 535,2005.
[10] Satyavarahala R, Purandhar K, Sashianth S,etal. Process for preparing losartan[P].US 200 602 005,2006.
[11] Aslan T, Bicer T, Gulkok Y,etal. Process for preparing biphenyl-tetrazole compound[P].WO 2 006 098 705,2006.
[12] 何敬文. 药物合成反应(第1版)[M].北京:中国医药科技出版社,1995.
猜你喜欢
健康体检与管理(2022年2期)2022-04-15
中国药学药品知识仓库(2022年5期)2022-04-11
云南化工(2021年7期)2021-12-21
有机氟工业(2021年3期)2021-09-15
潍坊学院学报(2016年6期)2016-04-18
癌变·畸变·突变(2016年3期)2016-02-27
合成化学(2015年2期)2016-01-17
中国卫生标准管理(2015年8期)2016-01-15
印刷技术·数字印艺(2015年10期)2015-12-10
化工进展(2015年3期)2015-11-11
