
具有除草活性的大环内酯类衍生物的定量构效关系
2010-11-30 10:56段红霞王瑞刚张建军董燕红梁晓梅吴景平王道全
物理化学学报 2010年4期
段红霞 王瑞刚 张建军 董燕红 梁晓梅 吴景平 王道全
(中国农业大学应用化学系,农业部农药化学与应用技术重点开放实验室,北京 100193)
具有除草活性的大环内酯类衍生物的定量构效关系
段红霞 王瑞刚 张建军 董燕红 梁晓梅*吴景平 王道全*
(中国农业大学应用化学系,农业部农药化学与应用技术重点开放实验室,北京 100193)
研究了一系列结构新颖的具有除草活性的大环内酯衍生物的定量构效关系(QSAR).构建的比较分子力场分析(CoMFA)、比较分子近似指数分析(CoMSIA)和全息定量构效关系(HQSAR)分子模型的交叉验证系数均大于0.5,非交叉验证系数r2都超过0.8,表明获取的QSAR模型具有可信的预测能力.对CoMFA、CoMSIA模型的三维(3D)等势图分析,发现除了立体场和静电场外,疏水场和氢键受体场也是影响大环内酯类化合物除草活性的重要因素.构建的HQSAR模型的原子贡献图提示的结构改造信息与三维QSAR的结果基本一致.利用CoMFA、CoMSIA模型提供的信息,对目前已合成的活性最高化合物B1-3进行分子结构改造,预测结果发现部分化合物可能具有更好的除草活性.
比较分子力场分析;比较分子近似指数分析;全息定量构效关系;大环内酯;除草活性
苯氧羧酸类除草剂的研究与开发始于上世纪40年代,2,4-二氯苯氧乙酸(2,4-D)是其成功的例子[1].其后,围绕2,4-D的分子结构开展的研究工作,促使更多具有更高除草活性或具有更广除草谱的化合物商品化[2].我们在新农药的创制研究中,模仿具有广谱生物活性的大环内酯类化合物的结构,将大环内酯环引入到芳氧羧酸类除草剂的分子结构中,合成了一系列结构新颖的含十六元环内酯的芳氧羧酸酯类化合物[2].生物测试结果表明,它们对单子叶植物和双子叶植物均具有较好的除草活性.为了探讨其进一步结构改造的可能性,研究其构效关系是必要的.在分子结构与生物活性的定量构效关系(QSAR)研究中,以80年代末发展起来的比较分子力场分析方法(CoMFA)[3]及在CoMFA方法基础上扩展起来的比较分子近似指数分析方法(CoMSIA)[4]最为有效,已成为药物和农药领域重要的研究工具.国内外一些研究小组在新农药的创制,特别是在除草剂的研究中,采用上述两种方法研究其构效关系,为进一步的分子设计提供参考,加速了创制的进程.研究的化合物类型主要包括:磺酰脲类[5],三唑并嘧啶磺酰胺类[6],氰基丙烯酸酯类[7],嘧啶苯甲酸类[8],咪唑啉酮类[9],异吲哚啉酮类[10],膦酸酯类[11],N-取代苄胺类[12],二苯醚类[13],单脒类[14].近年来,分子全息定量构效关系(HQSAR)法由于具有模型建立快速、预测能力高且能直接给出分子中对活性起主要作用的原子等优点成为一种新型高效的定量构效关系研究方法[15],目前HQSAR技术已经成功地应用到许多不同类化合物的构效关系研究当中[16-18].本文采用CoMFA、CoMSIA和HQSAR法研究前面提到的大环内酯类化合物的结构与对苋菜除草活性之间的定量构效关系,以便为进一步的结构改造提供参考.
1 研究方法
本文所有计算均使用SGI分子设计工作站上安装的Sybyl 7.3软件完成[19],计算中所选用的参数均为默认值.
1.1 目标化合物
本文研究的化合物及其对苋菜的除草活性均来源于文献[2],化合物的结构通式见图1,结构及除草活性如表1所示.

图1 研究化合物A,B及预测化合物P的结构通式Fig.1 Structural formula of the studied compounds A,B and predicted compounds P

表1 研究化合物的结构和对苋菜的除草活性(PLC50)Table 1 Structure and herbicidal activity(PLC50)to Amaranthus tricolor L.of the studied compounds
所研究化合物在结构上有三处变化:大环内酯环上C15上的取代基(R),侧链上的芳基(Ar),和羧酸部分α-C上的取代基(R1,R2),除草活性采用致死中浓度的对数(PLC50)表示.
所有化合物因含肟醚结构而存在顺反异构.文献[2]研究表明,对应的顺式异构体和反式异构体对苋菜的除草活性基本无差异,顺反异构体的除草活性等同于单一异构体的除草活性.基于上述事实,本文采用顺反异构体的生物测试数据表征顺式和反式构型化合物的各自除草活性,进行QSAR建模.
1.2 分子构象优化
分子构象的确定是建立有效3D-QSAR模型的首要前提.最好能获取各个活性化合物的药效构象,但由于通常化合物的靶标结构未知,很难确定它们真正的药效构象,通常采用分子的最低能量构象作为可能的药效构象进行理论研究.
大环内酯类化合物因含大环骨架结构具有较大的柔性,因此,我们选取已经报道的化合物A1-6的Z型异构体的晶体结构[2]为分子模板,利用单晶数据中大环母体骨架数据矫正并优化其它分子结构,进而使用Sybyl 7.3程序包做进一步结构优化.通过对计算方法、分子力场、电荷参数的系统考察,确定适用于大环内酯类化合物的最佳优化参数包括:方法, Broyden-Fletcher-Goldfarb-Shanno(BFGS);收敛梯度,0.21 kJ·mol-1·nm-1(即为0.005 kcal·(mol·Å)-1);力场,MMFF94;电荷,MMFF94,其它参数选取默认值.在上述优化参数的条件下,获得Z型A1-6分子构象较接近其单晶结构,如图2所示.
1.3 分子叠加
CoMFA和CoMSIA方法均是在分子周围三维网格上建立相关分子场,分析其与生物活性的相关性,网格点上能量的大小与分子的相对位置密切相关.因此,确定分子的取向是CoMFA和CoMSIA研究的关键步骤之一.所有分子需要选取一定的定位规则在空间叠合,才能保证分子场取向的一致性.目前广泛采用的定位方式是fit atoms,即以较高活性的分子为模板,选定基本骨架相似的原子,使其它要定位分子的相应原子与模板中原子的距离均方根偏差(RMSD)最小,此方法又称atoms-by-atom[20].

图2 A1-6的Z型晶体结构[2](a)与优化后的构象(b)Fig.2 Crystal structure[2](a)and optimizedconformation(b)of Z-A1-6

图3 E-A1(a)与Z-A1(b)的大环骨架Fig.3 Skeletons of macrocycle of E-A1(a)and Z-A1(b)
我们分别选取活性较好、具有一定代表性的A1-11的Z型和E型异构体为模板,以大环骨架及肟酯结构为叠合基本骨架(图3),运用Sybyl 7.3中的alignment database模块进行自动叠合,以保证所有分子在三维网格中的取向具有一致性,使分子间相互重叠时均方根偏差最小.叠合结果如图4所示,所有分子的叠合效果是令人满意的.
1.4 CoMFA和CoMSIA模型建立
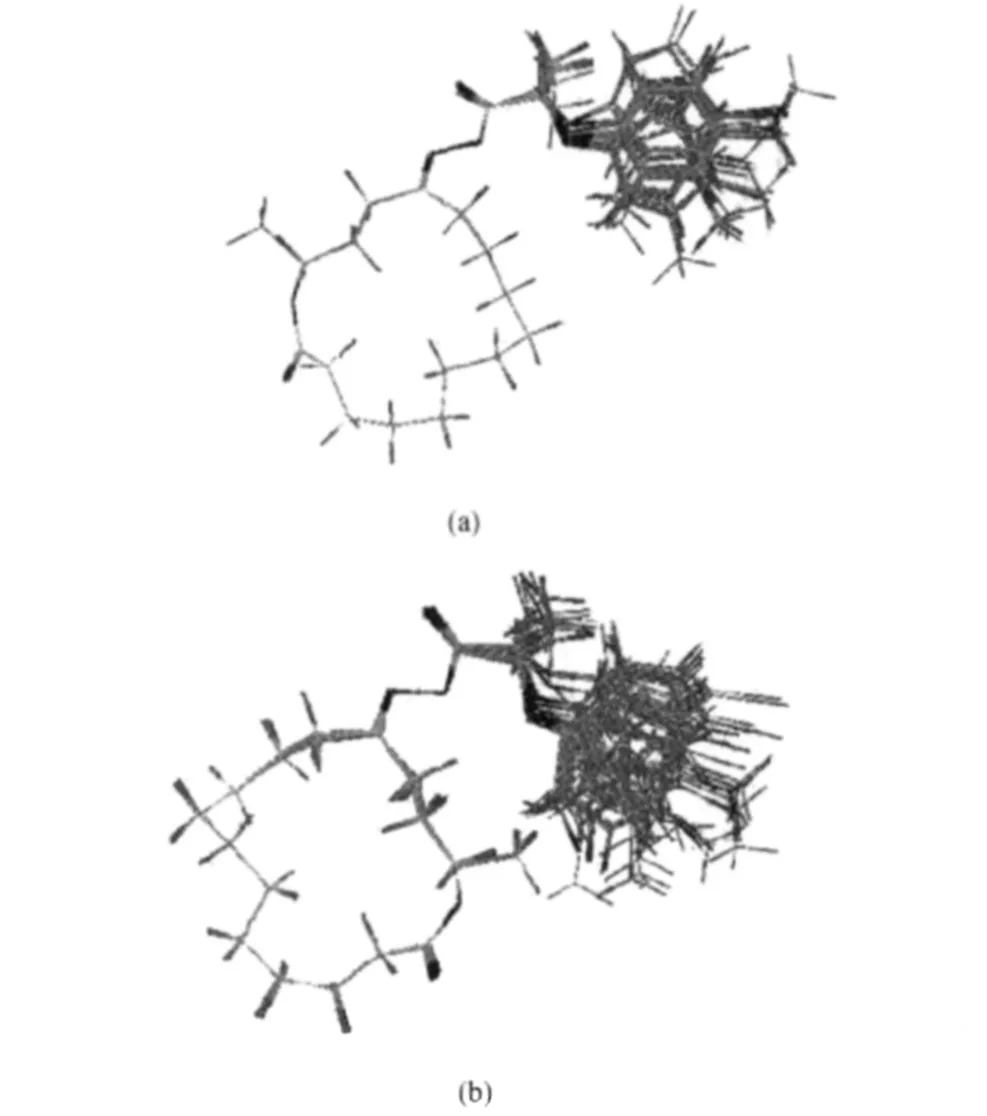
图4 研究化合物的E-系列(a)与Z-系列(b)分子叠合图Fig.4 Maps of superimposition of E-series(a)and Z-series(b)
所有CoMFA和CoMSIA研究均在Sybyl 7.3的QSAR模块中进行.选取sp3杂化的C+作为分子探针,对叠合分子外围网格点上的各种场能大小和分布进行计算.网格点步长设置为0.2 nm,场能的截断值设置为125.4 kJ·mol-1,柱过滤值设置为8.36 kJ·mol-1.在CoMFA研究中考虑立体场(steric)和静电场(electrostatic);在CoMSIA计算中引入立体场、静电场、疏水场(hydrophobic)、氢键受体场(acceptor).在PLS分析时,首先采用抽一法(LOO)进行交叉验证,求得交叉验证系数用来衡量模型的预测能力,一般认为,当时模型具有可信的预测能力,可继续进行非交叉验证),然后通过确定的最优主成分数进行非交叉验证回归分析,得到非交叉验证系数r2,建立相应的3D-QSAR模型[21].最后,采用Stedev coeff方法[22]显示所构建的3D-QSAR模型的三维等势图,直观地反映各个分子场对化合物生物活性的贡献.通过对训练集分子和新设计的化合物的活性预测,评价所建3D-QSAR模型的预测能力.
1.5 HQSAR模型的建立
分子全息结构-活性关系技术(HQSAR)是将系列化合物的分子结构转换为分子碎片数目,然后用偏最小二乘回归方法将分子碎片数目的类型即分子全息与生物活性之间建立相关关系.HQSAR方法只要求输入分子的二维结构和生物活性数据,分子碎片和全息的产生完全是自动的,非常迅速.HQSAR方法不但避免了经典QSAR方法的共线性和参数计算与选择的问题,而且不需要面对3D-QSAR中难以确定的叠合规则困难,具有极大的优点和广泛的应用前景.
采用Sybyl 7.3软件在SGI工作站上进行分子模拟,HQSAR模块产生分子全息.首先,每个分子的结构被划分为包含一定原子个数的分子碎片,其中,碎片中包含的原子数目被定义为碎片大小参数;碎片中所包含的结构类型有原子、键、连接方式、氢和手性等.根据碎片中包含原子数目的多少,分为较小原子碎片(1-3个原子),中等原子碎片(4-7个原子)和较大原子碎片(3-10个原子)三种情况.然后,对应于一个分子的所有亚结构碎片按照CYC算法[23]映射成一定长度的整数串,这即分子全息.整数串的长度称为分子全息长度参数,在HQSAR模块中,提供12个缺省的质数(53,59,61,71,83,97,151,199, 257,307,353和401)作为全息长度.通过调整碎片类型、碎片长度以及全息长度等参数,改善模型的统计能力.
采用PLS进行建模和交叉验证.分别以“逐一剔除”和“分组剔除”的交叉验证相关系数平方值来反映模型的稳健性和预测能力,用非交叉验证相关系数平方值r2和标准估计误差SEE表征模型的拟合程度.采用不同颜色对分子的各个原子进行编码,以反映各原子对生物活性贡献的大小,从而快速指导新配体的合成.
2 结果与讨论
2.1 CoMFA模型分析与讨论
我们对所选取的29个化合物进行CoMFA模型构建,计算结果如表2所示.获得CoMFA模型的统计学参数为:对于E系列化合物,其交叉验证相关系数,对Z系列化合物,得到的交叉验证系数,表明模型具有较好的可靠性和预测能力;由最佳主成分数确定的CoMFA模型的非交叉验证系数分别为r2=0.948(E系列),r2=0.944(Z系列);对于E系列化合物,立体场和静电场对模型的贡献分别为62.7%和37.3%,Z系列化合物的立体场和静电场对模型的贡献分别为63.9%和36.1%,可见,无论是E系列还是Z系列,立体场的贡献明显大于静电场,说明立体场对大环内酯类化合物与其靶标的相互作用影响更为显著.通过上述一系列的研究结果表明,尽管E系列与Z系列化合物在结构上有一定差异,但其生物活性具有很大的相似性,这与实测结果是一致的[2].
利用建立的CoMFA模型对部分训练集分子进行除草活性预测,评价模型的实际预测能力,计算结果如表3所示.可见,无论是E构型还是Z构型, CoMFA模型对大环内酯类化合物除草活性的预测结果与实验值吻合度都较高,差值在误差允许范围内,这表明构建的3D-QSAR模型具有很好的重现性和预测能力,能够进一步对同类其它大环内酯类化合物的除草活性进行预测.
CoMFA方法由于使用的自变量远大于因变量,故难以用数学函数表示,多采用直观的三维图形表示3D-QSAR模型的计算结果,从中分析化合物各个取代基性质及位置变化对活性的影响,获取系列化合物结构改造的信息.为了作出更好说明,选择具有最高生物活性的化合物B1-3作为参考分子,采取Stedev coeff方法获得CoMFA模型分子场的三维等势图(图5).在立体场等势图中,绿色区域表示在该区域存在体积大的基团有利于活性的提高,黄色区域表示在该区域不宜引入位阻大的基团;在静电场等势图中,红色区域表示引入电负性基团有利于提高生物活性,蓝色区域表明引入正电性基团有利于生物活性的提高.
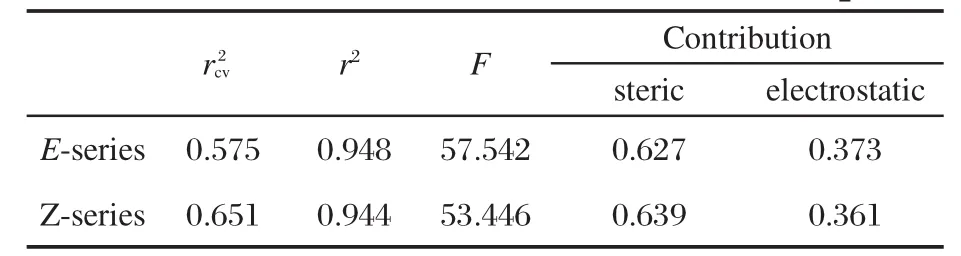
表2 E、Z两个系列化合物CoMFA建模结果Table 2 CoMFA models of E-and Z-series compounds
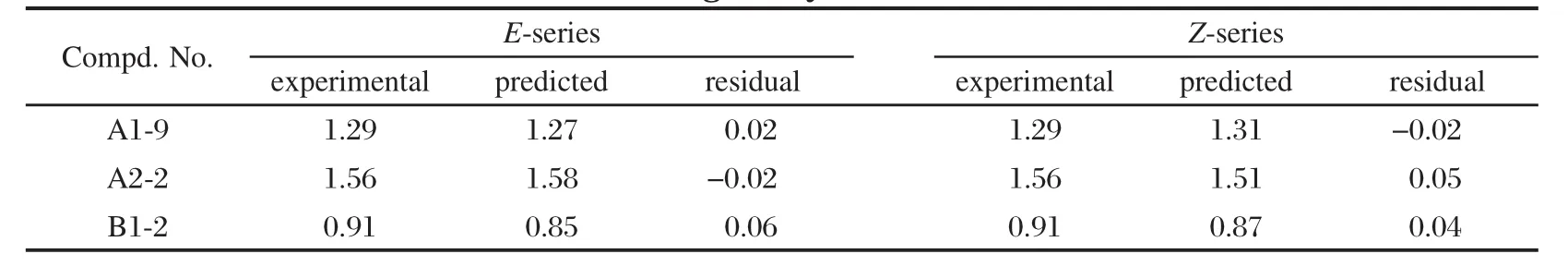
表3 部分训练集分子的除草活性(PLC50(mg·L-1))及采用CoMFA模型预测结果Table 3 Experimental and predicted herbicidal activity results(PLC50(mg·L-1))for some compounds in the training set by CoMFA model
如图5所示,就E系列化合物而言,在大环内酯骨架的C15位上,覆盖有较大范围的黄色区域,表明此位置不宜引入空间位阻较大的基团.我们正是在大环内酯C15位引入了较小的甲基(R=Me)合成了系列化合物,有效地增加了化合物的除草活性.如表1所示,B系列化合物的活性明显高于A系列化合物.同样,在侧链羧酸的α-位上也有黄色区域分布,说明这里也不宜引入体积过大的基团,如化合物A2-1(PLC50=1.35 mg·L-1)和化合物A3-1(PLC50= 1.54 mg·L-1),在该C原子上由一个甲基取代变成两个甲基后,化合物活性明显降低;此外,在苯环的对位覆盖有明显的绿色区域,说明在该位置引入空间位阻较大的基团有利于大环内酯类化合物活性的提高.如化合物A1-2(PLC50=1.74 mg·L-1)和化合物A1-3 (PLC50=1.69 mg·L-1)对比发现,增大苯环对位取代基的体积,即甲基替换为甲氧基后,化合物的活性有一定提高.可见,在苯环对位较大的取代基对生物活性是有利的.
图5还显示,在苯环4位上覆盖较大范围的蓝色区域,说明在该位置引入正电基团有利于大环内酯类化合物除草活性的提高.如对比化合物A1-1 (PLC50=1.86 mg·L-1)和A1-2(PLC50=1.74 mg·L-1)发现,苯环对位为带正电的甲基取代的化合物A1-2的活性要高些.此外,在苯环平面的上、下方覆盖大面积的红色区域,说明增加苯环的电负性会显著提高化合物的生物活性.如化合物A1-1(PLC50=1.86 mg·L-1)和化合物A1-11(PLC50=1.23 mg·L-1),由于吸电子基团—Cl,—Br的存在使得化合物A1-11的活性更高.对比图5中Z系列的CoMFA模型与E系列的CoMFA模型,发现两个系列的CoMFA模型图像提供的结构改造信息基本一致,这进一步论证了该类化合物的顺反异构对除草活性的影响很小.
2.2 CoMSIA模型分析与讨论
对叠加好的E、Z两个系列的化合物进行CoMSIA建模.在构建CoMSIA模型过程中,考虑了立体场、静电场、疏水场和氢键受体场,经过参数不断调整与优化,确定最佳的3D-QSAR模型.所有计算结果显示,构建的CoMSIA模型,说明具有较高的可信度.
为了作更好的解释说明,选择具有最高生物活性的化合物B1-3为参考分子,采取Stedev coeff方法获得CoMSIA模型各个分子场的三维等势图.所构建的CoMSIA模型的立体场和静电场等势图(略)与前述分析讨论的CoMFA模型的立体场和静电场所提供的化合物结构改造信息基本一致,这里不再累述.如图6所示,在疏水场等势图中,黄色区域表示该区域取代基疏水性增强有利于增加化合物的生物活性,灰色区域表示该区域疏水性减弱将提高化合物的生物活性.对E系列化合物,芳香环上方覆盖较大范围的黄色区域,这说明芳香苯环的疏水作用对大环内酯类化合物的除草活性极其重要,增加苯环的疏水性质,尤其是在2,3,5位增加取代基的疏水特征,可以有效的提高生物活性.在苯环4位分布有灰色区域,如若降低其取代基的疏水性质有可能增加化合物的生物活性.Z系列化合物疏水场提供的结构信息与E系列基本一致.

图5 E系列(a)和Z系列(b)CoMFA模型的立体场和静电场等势图Fig.5 Contour maps of steric and electrostatic fields for CoMFA models of E-series(a)and Z-series(b)
在氢键受体场等势图中,红色区域表示增大氢键受体场有利于提高活性,紫色区域表示减小氢键受体场有利于化合物活性的提高.如图7所示,无论是E系列化合物还是Z系列化合物,在大环内酯羰基邻位和侧链肟酯羰基附近均分布有较大范围的红色区域,如若在该位置引入具有氢键受体特征的基团将有利于增加大环内酯类化合物的生物活性;而在大环内酯羰基部位、侧链肟酯的N原子上覆盖有一定范围的紫色区域,此处不宜引入氢键受体基团.

图6 E系列(a)和Z系列(b)CoMSIA模型的疏水场等势图Fig.6 Contour maps of hydrophobic field for CoMSIA models of E-series(a)and Z-series(b)
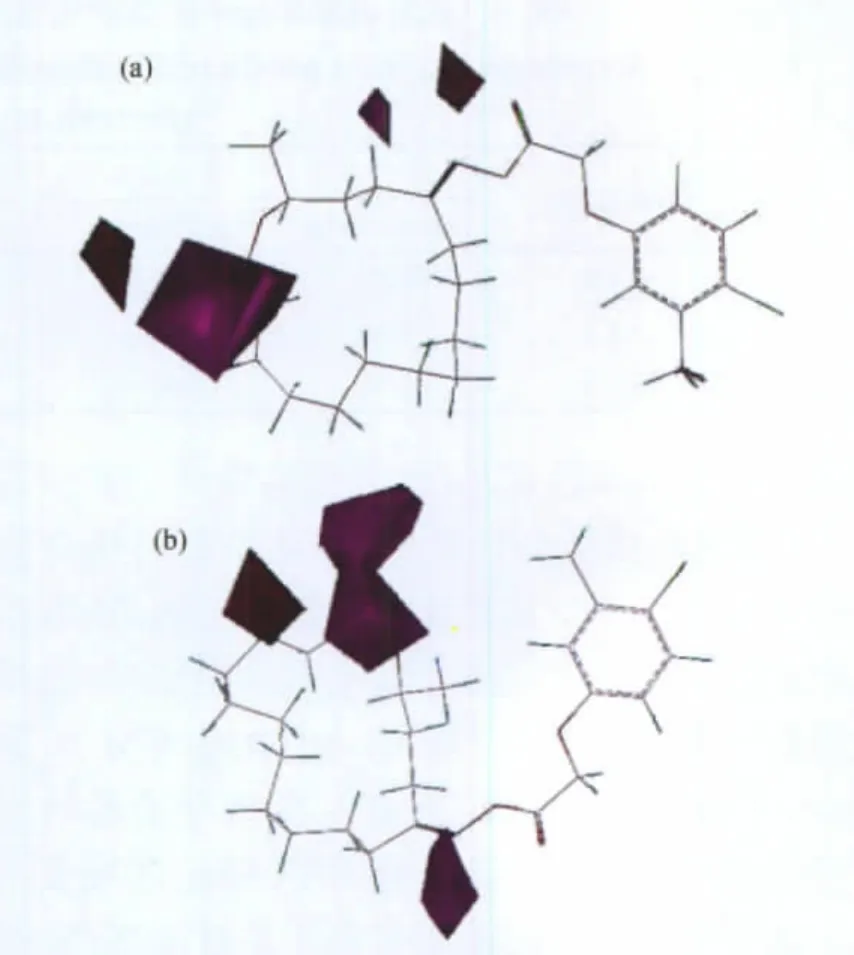
图7 E系列(a)和Z系列(b)CoMSIA模型的氢键受体场等势图Fig.7 Contour maps of hydrogen-bond acceptor field for CoMSIA models of E-series(a)and Z-series(b)
基于上述一系列的CoMFA和CoMSIA模型的研究结果,结合大环内酯类化合物的结构特征,我们发现除了立体场、静电场以外,疏水场和氢键受体场也是影响大环内酯类化合物除草活性的重要因素.
2.3 HQSAR模型分析与讨论

图8 A2-6的HQSAR模型的色码图Fig.8 Color coding figure of HQSAR model of A2-6

表4 不同碎片长度得到的HQSAR模型aTable 4 Influences of various fragment size parameters on HQSAR modela
分别在碎片长度为1-3、4-7和8-10范围内产生分子全息,得到一系列QSAR模型,列于表4.从模型“逐一剔除”的交叉验证结果来看,最佳模型处于分子碎片长度为8-10范围内,对应分子全息长度为97,交叉验证相关系数r2cv为0.680,模型的最佳主成分数为4.这是因为较大的分子碎片长度才能更详尽地区分大环内酯类化合物分子的不同结构特征.为了考察不同类型的碎片区分因素对模型质量的影响,在分子碎片长度为8-10、分子全息长度为97的条件下,改变所用的分子碎片区分参数例如原子类型A、化学键类型B、连接性Con、手性Ch、氢键给体和受体DA、氢原子H等,得到一系列的分子全息模型列于表5中.研究结果发现当考虑上述所有分子碎片区分参数组合时获得的模型质量最好,该HQSAR模型的统计学参数为0.713,最小标准预测偏差SEP为0.204;非交叉验证相关系数r2为0.870,标准估计误差SEE为0.137,最佳主成分数为4.一般认为,模型具有显著的预测能力.可见,所有分子碎片区分参数都对大环内酯类衍生物的除草活性有影响,尤其是氢键特征对生物活性的贡献最大,它的引入可以较大程度的提高HQSAR模型质量.
HQSAR的最大优点是可以采用不同颜色对分子中的各原子进行编码,以反映各原子对生物活性贡献的大小,从而快速直接地指导新配体的合成.处于光谱中红色系的颜色反映的是不利贡献,从橙色、橙红色到红色这种不利影响依次增大,合成时,应尽量避免引入这些基团.处于光谱蓝色系的颜色反映的是有利贡献,从黄色、蓝绿色到绿色表示对活性的贡献依次增大,在合成时这些基团应尽量保留.而白色,则表示对活性没有影响.
以化合物A2-6为例对HQSAR模型进行色码解释,如图8所示.图中青色为该系列化合物的共同结构.A2-6化合物R1位甲基上的氢原子呈现绿色或黄色,表明它们对化合物的活性有正性贡献,而R2位置的氢原子呈现白色对活性没有影响.所以在化学合成过程中应该尽可能保留R1位甲基取代,这一研究结果与之前CoMFA和CoMSIA模型中所指出的R1取代基体积不能过大的结论是一致的.其原因可能是只有合适大小的取代基如一个CH3,才能与靶标有较好的结合作用;若R1和R2同时取代,会导致空间位阻过大,不利于与受体很好的作用.取代基R位置上的氢原子呈现橙红色,表明该位置原子对活性的贡献是不利的,应该考虑引入新的取代基团.我们实验结果已经证实,R位置被甲基取代后化合物活性有显著提高(如化合物B1-2的活性为PLC50=0.90 mg·L-1,化合物A1-9的活性为PLC50=1.29 mg·L-1).苯环3位被取代的原子呈现黄色或绿色,表明该位置取代对活性是有利的,在合成过程中应尽量保留3位的取代,实验结果已经充分证实了这一点(如化合物B1-2的活性为PLC50=0.90 mg·L-1,化合物B1-3的活性为PLC50=0.53 mg·L-1). 4位氢原子呈现红色,说明对活性是不利的,因此在具体的合成过程中应该考虑设计其它基团取代,这与CoMFA和CoMSIA模型中指出的在苯环对位引入具有特性的取代基团对该系列化合物的除草活性是非常有利的结论相吻合,我们在实验过程中已经充分考虑到这一点,基本上设计的都是在4位引入不同取代基团来提高系列化合物活性.与肟酯中N相邻的大环内酯上的氢原子呈现绿色,对活性正性贡献,其它距离较远的氢原子为红色或橙色对活性负性贡献.

表5 碎片区分因素对HQSAR模型质量的影响aTable 5 Influences of various fragment type parameters on HQSAR modela
2.4 新分子活性预测
结合上述构建的CoMFA、CoMSIA模型的研究结果,以目前合成的大环内酯类化合物中活性最好的B1-3(PLC50=0.53 mg·L-1)为母体,分别对大环母体、侧链及侧链苯环上取代基进行结构改造,运用CoMFA模型对其除草活性进行预测,期望能获取具有更高除草活性的新型分子.设计的部分新化合物结构及其预测的PLC50值如表6所示.
结合CoMFA模型的立体场等势图信息,对大环内酯骨架部分的羰基和甲基进行结构改造,将B1-3大环内酯骨架上的—CH3换成—F、—CF3、—CH2CH3、—OCH3、—CN后,预测活性有所降低(如P1),这进一步证明保留大环内酯骨架上的—CH3对活性是有利的;考虑CoMSIA模型的氢键受体场等势图结果,大环部分的羰基作为氢键受体不应削弱,当羰基变为硫羰基后,预测活性明显降低(如P2).
根据CoMFA模型的立体场和静电场信息,对B1-3侧链羰基上的α-CH2进行结构改造.将两个—H中的一个用强电负性的—F取代,活性有所提高(如P3);但将两个—H均由—F取代后,可能由于空间位阻的原因,活性下降(如P4).
对B1-3苯环部分的改造:根据CoMFA模型的立体场和静电场等势图结果,结合CoMSIA模型的疏水场等势图信息,保持对位Cl原子不变,在邻、间位上引入空间位阻大、或电负性强、或疏水性强的基团,如—CN、—CF3、苯基、环己基、环丙基等,结果发现只有在间位上单一引入这些取代基团,生物活性才有望提高(如P7、P8、P9、P10、P11).可见,在以后的结构改造中应着重针对苯环间位进行结构改造.此外,在保留间位CH3的情况下,将对位上的Cl原子替换为Br原子后预测活性有所下降(P5);但将其替换为电负性更强的NO2后,预测活性显著提高(P6),这可能是因为—NO2疏水性弱的缘故.同时将侧链羧酸α-C上的—H替换为—F及苯环对位Cl替换成NO2,同样可以获取良好的效果,活性有很大的提高(如P13、P14).因此,在后续的结构改造过程中,建议在侧链羧酸α-C上引入F,在苯环对位引入NO2,而间位则引入体积更大、给电子和疏水性均强的烃基等基团,有望获得除草活性更好的新型大环内酯类化合物.

表6 部分设计化合物的结构及除草活性的预测Table 6 Structure and predicted herbicidal activity of designed compounds
3 结 论
运用比较分子力场分析(CoMFA)、比较分子相似性指数分析(CoMSIA)和全息定量构效关系(HQSAR)方法构建了具有除草活性的大环内酯类系列化合物的定量构效关系模型.对部分已知化合物进行除草活性的预测结果与实验值相当,说明所构建的模型具有良好的预测能力.研究结果表明,由CoMFA和CoMSIA模型获得的结构改造信息基本一致,除了立体场和静电场外,疏水场和氢键受体场也是影响上述大环内酯类衍生物除草活性的重要因素.通过构建最优HQSAR模型给出的原子贡献图提示的化合物结构改造信息与三维QSAR提供的结果相吻合.根据构建的CoMFA和CoMSIA模型,以B1-3为母体,分别对大环内酯环,侧链及侧链苯环上的取代基进行结构改造,并对其除草活性进行预测,结果获得数个可能具有更高活性的化合物,为大环内酯类化合物通过结构改造获得活性更高的化合物奠定了基础,也为其它类型大环化合物的构效关系研究提供了借鉴.
Supporting Information Available: CCDC 689231 contains the supplementary crystallographic data for this paper. These data can be obtained free of charge from the CCDC,12 Union Road,Cambridge CB2 1EZ,UK;Tel:(44)01223 762910; E-mail:deposit@ccdc.cam.ac.uk.
1 Fletcher,W.W.;Kirkwood,R.C.Herbicides and plant growth regulators.London:Granada Publishing,1982:21-27
2 Meng,X.Q.;Zhang,J.J.;Liang,X.M.;Dong,Y.H.;Huang,J.X.; Rui,C.H.;Fan,X.L.;Chen,F.H.;Wang,D.Q.J.Agric.Food Chem.,2009,57(2):610
3 Cramer,R.D.;Patterson,D.E.;Bunce,J.D.J.Am.Chem.Soc., 1988,110:5959
4 (a)Klebe,G.;Abraham,U.;Mietzner,T.J.Med.Chem.,1994,37: 4130 (b)Klebe,G.;Abraham,U.J.Comput.-Aided Mol.Des., 1999,13:1 (c)Böhm,M.;Stürzebecher,J.;Klebe,G.J.Med.Chem.,1999, 42:458 (d)Bordäs,B.;Kömives,T.;Lopata,A.Pest Manag.Sci.,2003, 59:393
5 (a)Liu,J.;Li,Z.M.;Wang,X.;Ma,Y.;Lai,C.M.;Jia,G.F.; Wang,L.X.Sci.China Ser.B-Chem.,1998,41(1):50 (b)Hou,T.J.;Li,Z.M.;Li,Z.;Liu,J.;Xu,X.J.J.Chem.Inf. Comput.Sci.,2000,40(4):1002 (c)Wang,B.L.;Ma,N.;Wang,J.G.;Ma,Y.;Li,Z.M.;Li,Y.H. Acta Phys.-Chim.Sin.,2004,20(6):577 [王宝雷,马 宁,王建国,马 翼,李正名,李永红.物理化学学报,2004,20(6):577] (d)Ma,Y.;Jiang,L.;Li,Z.M.;Lai,C.M.Chem.J.Chin.Univ., 2004,25(11):2031 [马 翼,姜 林,李正名,赖城明.高等学校化学学报,2004,25(11):2031] (e)Ban,S.R.;Niu,C.W.;Chen,W.B.;Ren,X.B.;Yu,Z.H.;Xi, Z.Chem.J.Chin.Univ.,2007,28(3):543 [斑树荣,牛聪伟,陈文彬,任晓白,余志红,席 真.高等学校化学学报,2007,28(3): 543] (f)Yu,Z.H.;Niu,C.W.;Ban,S.R.;Wen,X.;Xi,Z.Chin.Sci. Bull.,2007,52(14):1929 (g)Guo,W.C.;Ma,Y.;Li,Y.H.;Wang,S.H.;Li,Z.M.Acta Chim.Sin.,2009,67(6):569 [郭万成,马 翼,李永红,王素华,李正名.化学学报,2009,67(6):569]
6 (a)Ren,T.R.;Chen,H.M.;Xie,G.R.;Zhou,J.J.;Chen,F.H. Chem.J.Chin.Univ.,1998,19(12):1950 [任天瑞,陈红明,谢桂荣,周家驹,陈馥衡.高等学校化学学报,1998,19(12):1950] (b)Yang,G.F.;Liu,H.Y.;Yang,X.F.;Yang,H.Z.Acta Chim. Sin.,1999,57(7):706 [杨光富,刘华银,杨秀风,杨华铮.化学学报,1999,57(7):706] (c)Yang,G.F.;Liu,H.Y.;Yang,X.F.;Yang,H.Z.Acta Phys.-Chim.Sin.,1999,15(2):190 [杨光富,刘华银,杨秀风,杨华铮.物理化学学报,1999,15(2):190]
7 (a)Liu,X.L.;Sun,M.;Wen,X.;Qi,L.N.;Miao,F.M.Comput. Appl.Chem.,2000,17(1-2):11 [刘小兰,孙 命,文 欣,齐丽宁,缪方明.计算机与应用化学,2000,17(1-2):11] (b)Han,X.F.;Liu,Y.X.;Liu,Y.;Lai,L.H.;Huang,R.Q.;Wang, Q.M.Chin.J.Chem.,2007,25(8):1135 (c)Liu,Y.X.;Wei,D.G.;Zhu,Y.R.;Liu,S.H.;Zhang,Y.L.; Zhao,Q.Q.;Cai,B.L.;Li,Y.H.;Song,H.B.;Liu,Y.;Wang,Y.; Huang,R.Q.;Wang,Q.M.J.Agric.Food Chem.,2008,56(1): 204
8 (a)Li,A.X.;Wang,J.L.;Su,H.Q.;Sun,M.;Yu,M.;Miao,F.M. Comput.Appl.Chem.,2000,17(1-2):27 [李爱秀,王谨玲,苏华庆,孙 命,郁 铭,缪方明.计算机与应用化学,2000,17(1-2): 27] (b)Su,H.Q.;Wang,J.L.;Li,A.X.;Sun,M.;Miao,F.M.Chem. Res.Appl.,1999,11(6):626 [苏华庆,王谨玲,李爱秀,孙 命,缪方明.化学研究与应用,1999,11(6):626]
9 Wang,J.L.;Li,A.X.;Su,H.Q.;Sun,M.;Miao,F.M.Acta Chim. Sin.,1999,57(12):1291 [王谨玲,李爱秀,苏华庆,孙 命,缪方明.化学学报,1999,57(12):1291]
10 Soung,M.G.;Lee,Y.J.;Sung,N.D.Bull.Korean Chem.Soc., 2009,30(3):613
11 Peng,H.;Wang,T.;Xie,P.;Chen,T.;He,H.W.;Wan,J.J.Agric. Food Chem.,2007,55(5):1871
12 Feng,X.;Yao,J.H.;Lü,L.;Tang,Q.H.;Fan,B.T.Acta Chim. Sin.,2006,64(11):1097 [冯 骁,姚建华,吕 龙,唐庆红,范波涛.化学学报,2006,64(11):1097]
13 Wang,J.L.;Li,A.X.;Yu,M.;Zhai,X.H.;Miao,F.M.Comput. Appl.Chem.,2000,17(1-2):25 [王谨玲,李爱秀,郁 铭,翟秀红,缪方明.计算机与应用化学,2000,17(1-2):25]
14 Wang,B.L.;Wang,J.G.;Ma,Y.;Li,Z.M.;Li,Y.H.;Wang,S.H. Acta Chim.Sin.,2006,64(13):1373 [王宝雷,王建国,马 翼,李正名,李永红,王素华.化学学报,2006,64(13):1373]
15 HQSAR is the product of Tripos Inc.,St.Louis,MO,1997
16 Avery,M.A.;Alvim-Gaston,M.;Woolfrey,J.R.J.Med.Chem., 2002,45:292
17 Li,H.;Zhang,H.B.Acta Chim.Sin.,2005,63(11):1018 [李 华,张华北.化学学报,2005,63(11):1018]
18 Wang,X.D.;Xiao,Q.F.;Cui,S.H.;Liu,S.S.;Yin,D.Q.;Wang, L.S.Sci.China Ser.B-Chem.,2005,35(1):58 [王晓栋,肖乾芬,崔世海,刘树深,尹大强,王连生.中国科学B辑:化学,2005,35 (1):58]
19 SYBYL 7.3 is available from Tripos Associates Inc.,1699 S. Hanley Rd.,St.Louis,MO631444,USA
20 Xu,X.J.;Hou,T.J.;Qiao,X.B.;Zhang,W.Computer-aided drug design.Beijing:Chemical Industry Press,2004 [徐筱杰,侯廷军,乔学斌,章 威.计算机辅助药物分子设计.北京:化学工业出版社,2004]
21 Luo,X.Chemometrics.Beijing:Science Press,2001 [罗 旭.化学统计学.北京:科学出版社,2001]
22 Jiang,Y.R.;Qin,W.Acta Phys.-Chim.Sin.,2008,24(10):1859 [蒋玉仁,秦 伟.物理化学学报,2008,24(10):1859]
23 Masahiro,N.Toxicol.Lett.,1998,103:627
November 27,2009;Revised:January 25,2010;Published on Web:March 9,2010.
QSAR of Macrolactone Derivatives with Herbicidal Activity
DUAN Hong-Xia WANG Rui-Gang ZHANG Jian-Jun DONG Yan-Hong LIANG Xiao-Mei*WU Jing-Ping WANG Dao-Quan*
(Key Laboratory of Pesticide Chemistry and Application Technology,Ministry of Agriculture, Department of Applied Chemistry,China Agricultural University,Beijing 100193)
Westudiedthequantitativestructure-activityrelationships(QSAR)ofmacrolactone derivatives.Statistical results fromthe established comparative molecularfield analysis(CoMFA),the comparative molecular similarity indices analysis(CoMSIA),and the hologram quantitative structure-activity relationship(HQSAR)models showed believable predictability based on the cross-validated value(r2cv>0.5)and the non cross-validated value(r2>0.8).A contour maps analysis of the CoMFA and CoMSIA models showed that the hydrophobic and hydrogen-bond acceptor fields are important factors that affect the herbicidal activity of macrolactone compounds except for the steric and electrostatic fields.The structural modification information from the different atom contributions in the HQSAR model is in agreement with those of the 3D-QSAR models.According to the results from the CoMFA and CoMSIA models,the structure of compound B1-3 with the best herbicidal activity was modified and some designed compounds were predicted to have higher activity.
CoMFA;CoMSIA;HQSAR;Macrolactone;Herbicidal activity
O641
*Corresponding authors.Email:wangdq@cau.edu.cn,liangxiaomeim@yahoo.com.cn;Tel:+86-10-62732219.
The project was supported by the National Key Basic Research Program of China(973)(2003CB114407).
国家重点基础研究发展计划(973)(2003CB114407)资助项目
王道全,1978年10月考取北京大学分子工程与化学学院(原化学系)研究生,师从张滂院士,从事有机合成研究,1984年3月获理学博士学位.
猜你喜欢
分子催化(2022年1期)2022-11-02
中学化学(2022年5期)2022-06-17
高中数理化(2020年1期)2020-02-29
理科考试研究·高中(2019年8期)2019-09-19
中成药(2018年12期)2018-12-29
临床医药文献杂志(电子版)(2017年12期)2017-05-18
中南大学学报(自然科学版)(2016年2期)2017-01-19
浙江中西医结合杂志(2016年2期)2016-01-25
化学教学(2015年11期)2015-12-19
天然产物研究与开发(2014年7期)2014-04-27
