
PbmTen(m+n≤6)团簇的结构与稳定性
2010-12-12 02:45龚晓霞杜际广
物理化学学报 2010年6期
龚晓霞 杜际广 蒋 刚
(四川大学原子与分子物理研究所,成都 610065)
Lead telluride is a very important narrow-gap semiconductor, and it is widely used as infrared detector.It has some interesting characteristics,such as small energy gaps,low resistivities,large carrier mobilities,and unusually high dielectric constants.In recent years,more and more research interests are focused on its material properties for thermoelectric power generation.But for the lead,it is well known as the toxic metal which poses a great threat to the environment and human life,especially for children′s neurological and physical development.The lead compounds should be an important topic in the lead pollution.The study of lead telluride cluster might provide useful information for material investigate and pollution control.
There are many theoretical[1-6]and experimental[7-10]studies on the lead telluride.Nabi[11]and Hevine[12]et al.proved that lead telluride is narrow gap semiconductor.Lachhab et al.[13]used the full potential linearized augmented plane wave(LAPW)method to study the electronic structure of the PbTe compound. Grabecki et al.[14]pointed out that PbTe had broad application prospects in nanostructure physics.Mazzone[15]studied the fragmentation of Pb clusters with size N≤24 employing the timedependent Hartree-Fock method.Rajesh et al.[16]investigated the geometric and electronic structure of Pbn(n=2-15)clusters using the ab initio molecular dynamics simulation.Wang et al.[17]used DFT with Becke-Lee-Yang-Parr(BLYP)gradient correction to obtain the lowest-energy structures and electronic properties of the Pbn(n=2-22)clusters.Gingerich et al.[18]got the structure parameter of Pb2,Pb3and Pb4by mass spectrometry.Balasubramanian et al.[19]investigated the geometries and electronic states of Pb5. Liu et al.[20]studied the geometrical structures and stabilities of small PbmOn(m=1-4,n=1-2m)clusters using the hybrid B3LYP functional.Xing et al.[21]studied the PbnSnclusters(n=1-9)by using DFT method.The pure Te clusters have been studied extensively.Goddard[22]and Balasubramanian[23]et al.investigated the geometries,vibrational frequencies,and electronic characters of Te3by using DFT method.In the experiment,Hassanzadeh et al.[24]found the frequencies and low energy structures of Ten(n=2-4)clusters.Pan[25]obtained the geometric structures,electronic properties,and vibrational frequencies of tellurium clusters(n≤8)by using DFT method.The DFT method is widely used to investigate the structures and stabilities of the mixed cluster[26-27].
In spite of the abundant reports data above have been found, the investigations on the PbTe clusters are far from adequate.In order to understand the evolution of their physical and chemical properties with size,it is essential to study their stabilities and structural properties.In this article,heteronuclear PbmTen(m+n≤6)and homonuclear Pbn,Ten(n=2-6)clusters have been investigated using DFT[28-29]method.
1 Computational method
The present work is based on the Gaussian 03 package[30].The Becke three parameters hybrid functional with the Lee-Yang-Parr correlation functional(B3LYP)[31-32]has been adopted in our DFT calculation.Furthermore,the test computations with CEP-121G、LANL2DZ and SDD sets are performed for dimers(Pb2, Te2,and PbTe)to demonstrate which basis set is rational.The comparison results between the calculated values and experimental data[17,24,33]are given in Table 1.Note that the SDD basis set predicts bond lengths and frequencies of PbTe and Te2dimers to be slightly more close to experimental values.Therefore,the SDD employed in our calculation is successful to describe the clusters involving Pb,Te atoms.
During the geometry optimizations,to find the stable structures,the possible original structures and the spin states are systematically considered,the requested convergence on energy is 10-6a.u..In general,clusters with higher symmetry are expected to be more stable.Therefore,according to a structure with a known symmetry,the initial geometry is constructed by superposing the smaller building blocks.In addition,we can often simply add or subtract an atom from a cluster of size n to obtain geometry for a cluster of size n+1 or n-1.Moreover,we have consulted the geometric structure of pure Pb[16-17,26]clusters and Te[25,34]clusters.For mixed clusters,we have generated a numberof possible isomers that are adopted from the other semiconductor compound clusters like PbO[20]and ZnTe[34].
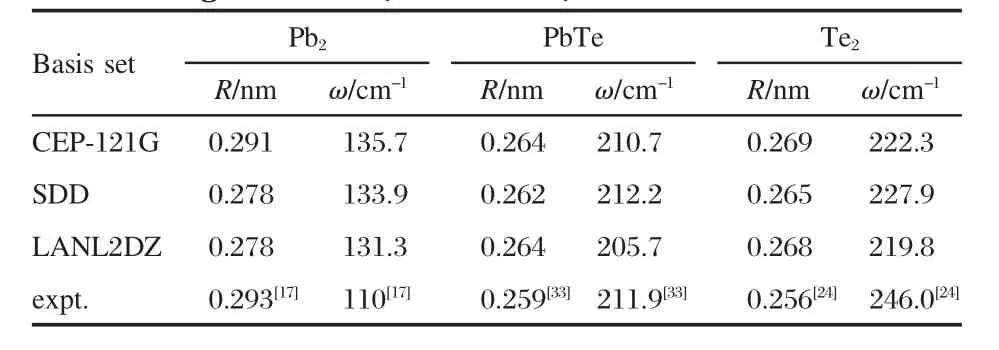
Table 1 Calculated bond length(R)and vibrational frequency(ω)for the ground state of Pb2,PbTe,and Te2 using CEP-121G,LANL2DZ,and SDD basis sets
The geometry optimization is performed without any symmetry constraints.The vibrational frequency calculations are also carried out to ensure whether the obtained structures correspond to local minima on the potential surface.The average binding energy per atom(Eb)is calculated by Eb=[E(PbmTen)-mE(Pb)-nE(Te)]/(m+n),where E are the total energies of corresponding systems.The dissociation energy is defined as ΔEd=EB+EC-EA, EB,EC,and EAare the total energies of B,C,and A clusters,respectively;it represents the dissociation channel that cluster A is dissociated into B and C congeries.
2 Results and discussion
2.1 Geometric structures
2.1.1 Homonuclear Pb clusters
The discussions on pure Pbnand Ten(n≤6)clusters will be simplified in the present paper due to our main attention on the mixed Pb-Te clusters.For the Pb2dimer,the present calculations predict the bond length and Ebto be 0.278 nm and 0.875 eV,respectively.They are found to be fitted well with experimental results(0.293 nm and 0.86 eV[17]).The global minimum structure of Pb3is an equilateral triangle with D3hsymmetry,the side length(0.303 nm)is close to the previous theoretical results (0.301 nm[16,26]).The ground state of Pb4cluster is a planar rhombus structure(D2h),the optimized side length(0.299 nm)is in agreement with previous theoretical results(0.298 nm[16],0.311 nm[17],0.304 nm[35]),Eb(1.81 eV)is a little larger than the calculation data(Eb=1.66 eV)obtained by Wang et al.[17].For Pb5cluster,the triangular bipyramid structure with D3hsymmetry is found to be the ground state,the side length(0.300 nm)is comparable with the theoretical results(0.297 nm[16],0.311 nm[17],0.30 nm[19]).In the case of Pb6,the crossed rhombus with D4hsymmetry is the lowest energy structure with the Pb—Pb distance of 0.310 nm.Rajesh et al.[16]predicted that the minimum energy structure is D4hsymmetry with the side length of 0.306 nm.
2.1.2 Homonuclear Te clusters
For Te2,we predict the bond length and the vibration frequency to be 0.265 nm and 227.9 cm-1,they are 0.009 nm larger for bond length and 12.1 cm-1lower for frequency with respect to the experimental results[24].For Te3cluster,the equilateral triangle structure with D3hsymmetry is the lowest energy structure, the Te—Te bond length(0.289 nm)is in accordance with the previoustheoreticalvalues(0.283nm[23]and0.28nm[22]).Themaximum vibrational frequency(200.9 cm-1)is also in accordance with the prediction value(198.5 cm-1)[34]and experiment result (232 cm-1)[24].The ground state of Te4cluster is the trapezoid structure with C2vsymmetry,and the maximum frequency(218.5 cm-1)is lower than the experimental result(233.9 cm-1)[24].The most stable structure of Te5cluster is an envelope structure with Cssymmetry,the shortest side length and the maximum frequency are 0.281 nm and 171.2 cm-1,respectively,theyare close to the theoreticalresult[34].For Te6cluster,we have found that the struc-ture with C2vsymmetry is the ground state structure.Pan[25]found that the lowest energy structure was a distorted hexagon with D3dsymmetry.The difference between the present results and Pan′s results could be due to the difference in basis set.
2.1.3 Heteronuclear PbTe clusters
The lowest-energy and low-lying structures of(PbTe)n(n=1-3)clusters are shown in Fig.1.For PbTe,the calculated bond length,vibrational frequency and the Ebare 0.262 nm,212.2 cm-1and 1.86 eV,respectively.In experiment,Huber and Herzberg[33]found the bond length of PbTe to be 0.259 nm.The global minimum structure of Pb2Te2is a rhombic structure with C2vsymmetry,and the structure is quite similar to the lowest-energy structure of Pb2O2cluster[20].For Pb3Te3,there are as many as thirteen stable structures,the distort hexagon structure with C2vsymmetry is the most stable one.
The Pb-rich clusters are shown in Fig.2(for PbnTe,n=2-5)and Fig.3(for PbnTe2,n=3-4).The ground state of the Pb2Te cluster is an acute isosceles triangle structure(C2v).The low-lying isomers have isosceles triangle structure with C2vsymmetry and linear structure with C∞vsymmetry.For Pb3Te,the butterly-like geometry structure with Cssymmetry is preferred cluster among all the low-lying isomers.The lowest-energy structure of Pb4Te is a capped bent rhombic structure(C2v).The ground state of Pb5Te is C4vsymmetry,and the other seven kinds of isomers have been shown.The most stable structure of Pb3Te2is similar to the Pb3Te,with adding bridged Te atom in the opposite site of the preexistent Te atom.For Pb4Te2,eight kinds of stable isomers have been gotten,the configuration of the lowest energy with Cssymmetry is similar to the ground-state structure of Pb4O2cluster[20].
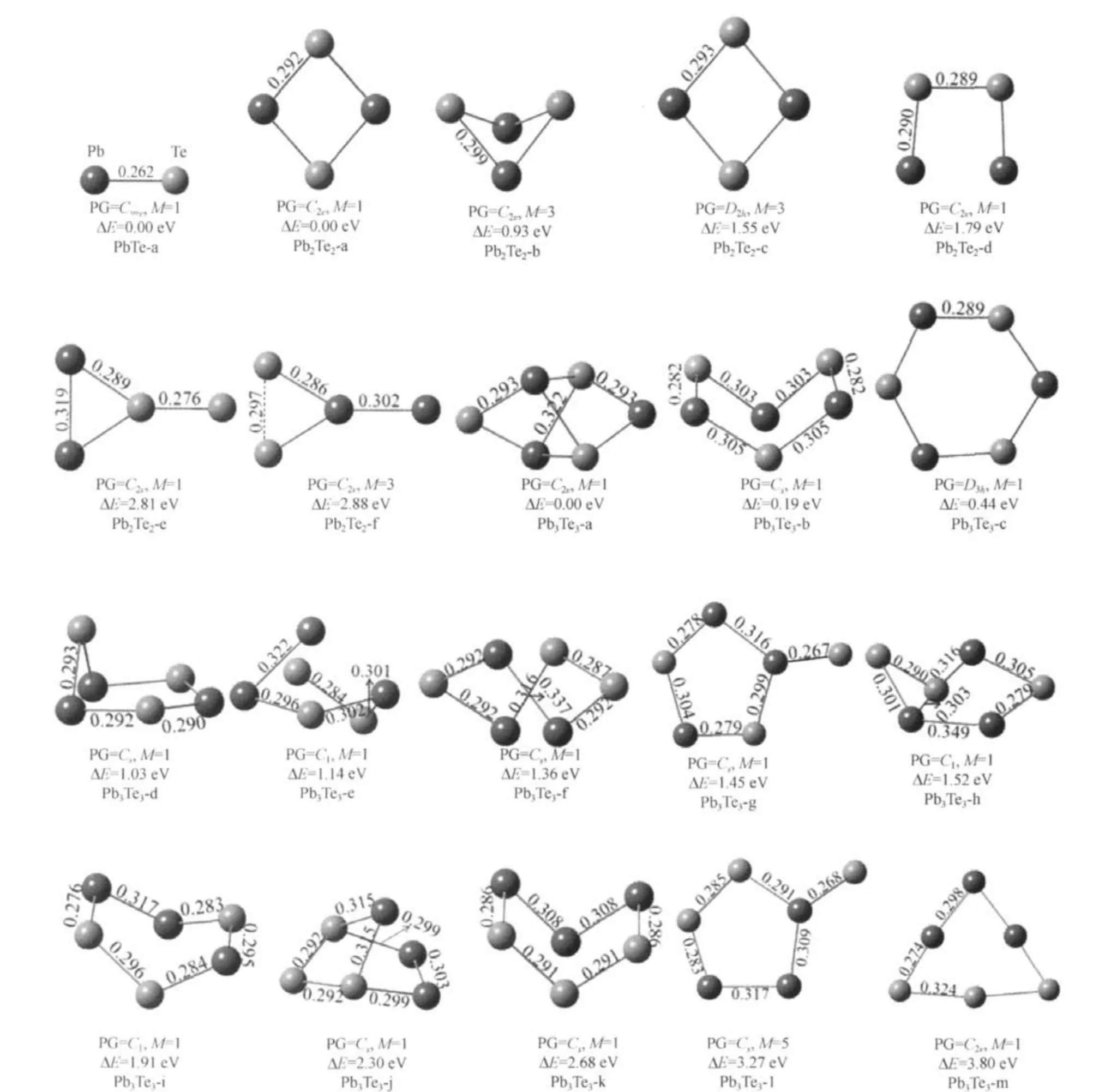
Fig.1 Stable geometries of(PbTe)n(n=1-3)clustersThe bond lengths(in nm),spin multiplicity(M),point group(PG)symmetry,and relative energy(relative to the ground state structures,ΔE)of these isomers are shown.
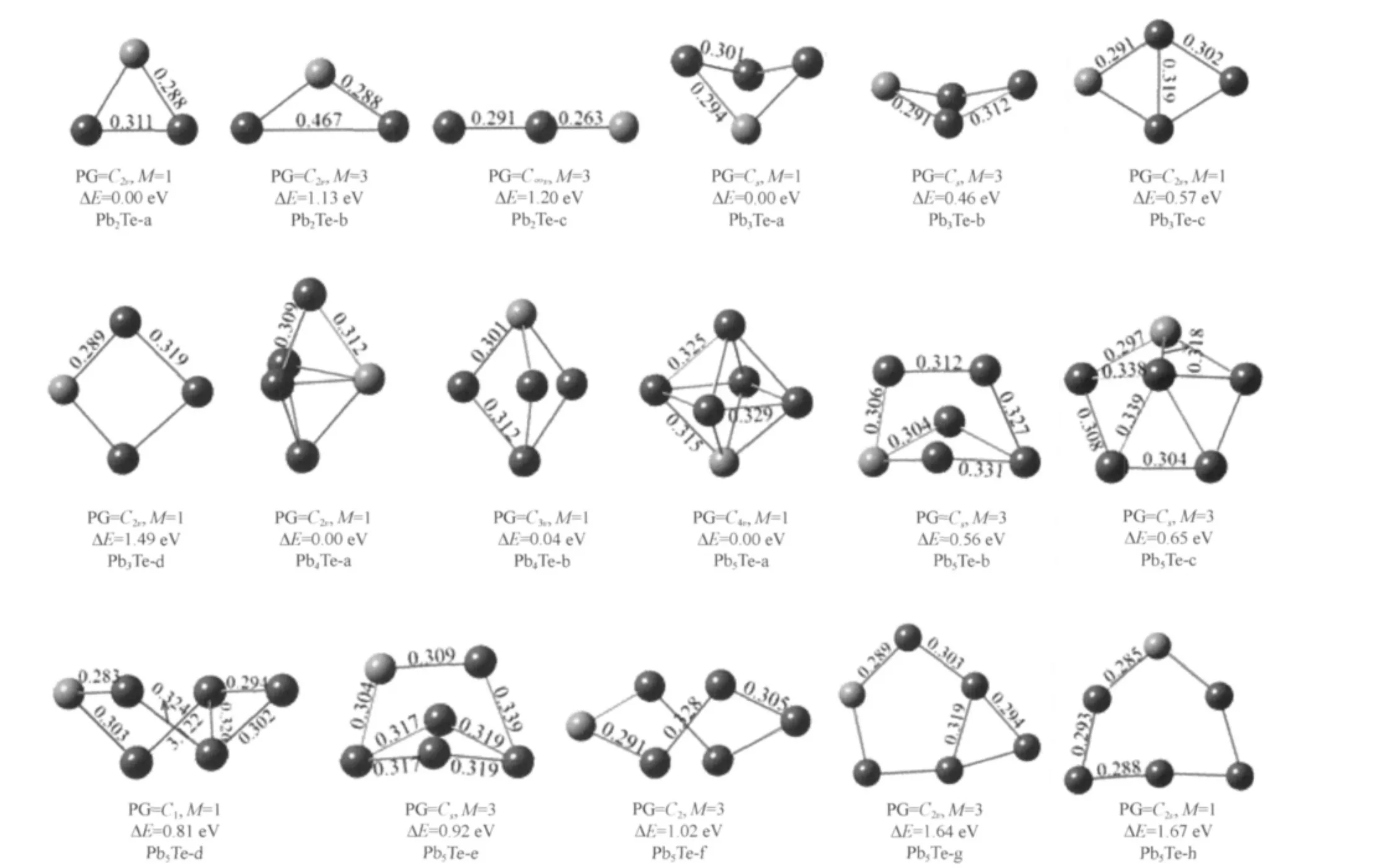
Fig.2 Stable geometries of PbnTe(n=2-5)clusters
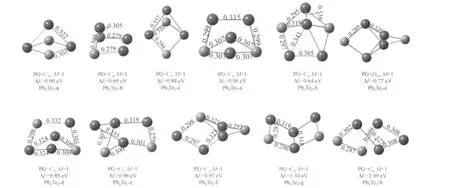
Fig.3 Stable geometries of PbnTe2(n=3-4)clusters
The stable structures of PbTen(n=2-5)and Pb2Ten(n=3-4)are shown in Fig.4 and Fig.5.For PbTe2,the most stable isomer is corresponding to an isosceles triangle structure with C2vsymmetry.The next stable linear TePbTe structure is 0.69 eV higher in energy than the most stable one.The lowest energy of PbTe3cluster is a plane geometry with C2vsymmetry,another stable structure with C1symmetry is found to be 0.2 eV higher in energy.The most stable structure of the PbTe4cluster is a pentagon structure with C2symmetry.For PbTe5,the lowest energy structure with C1symmetry is 0.13 eV lower in energy than another stable isomer(PbTe5-b).In the eight kinds of isomers,the pentagon structure(C2v)is found to be the ground state structure of Pb2Te3cluster.The ground state of Pb2Te4is the distort hexagon (Pb2Te4-a)structure with Cssymmetry.
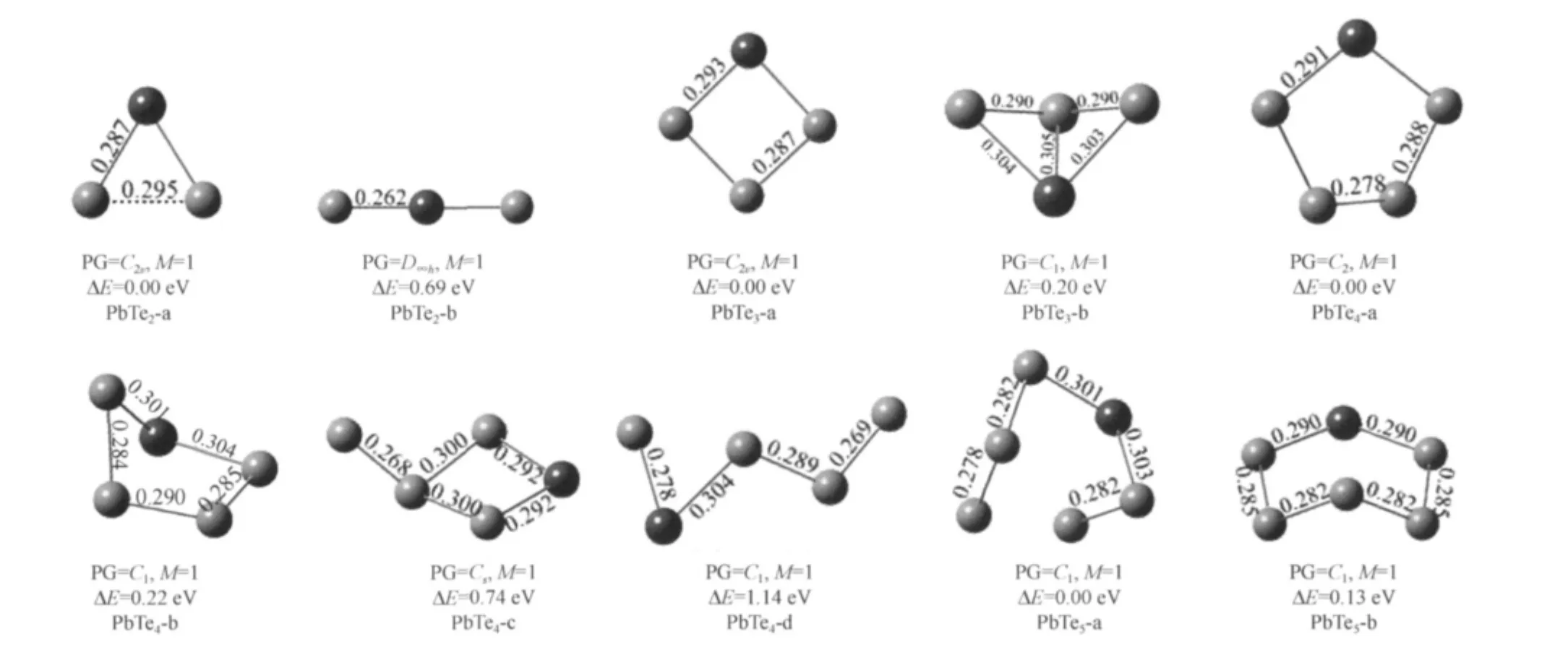
Fig.4 Stable geometries of PbTen(n=2-5)clusters
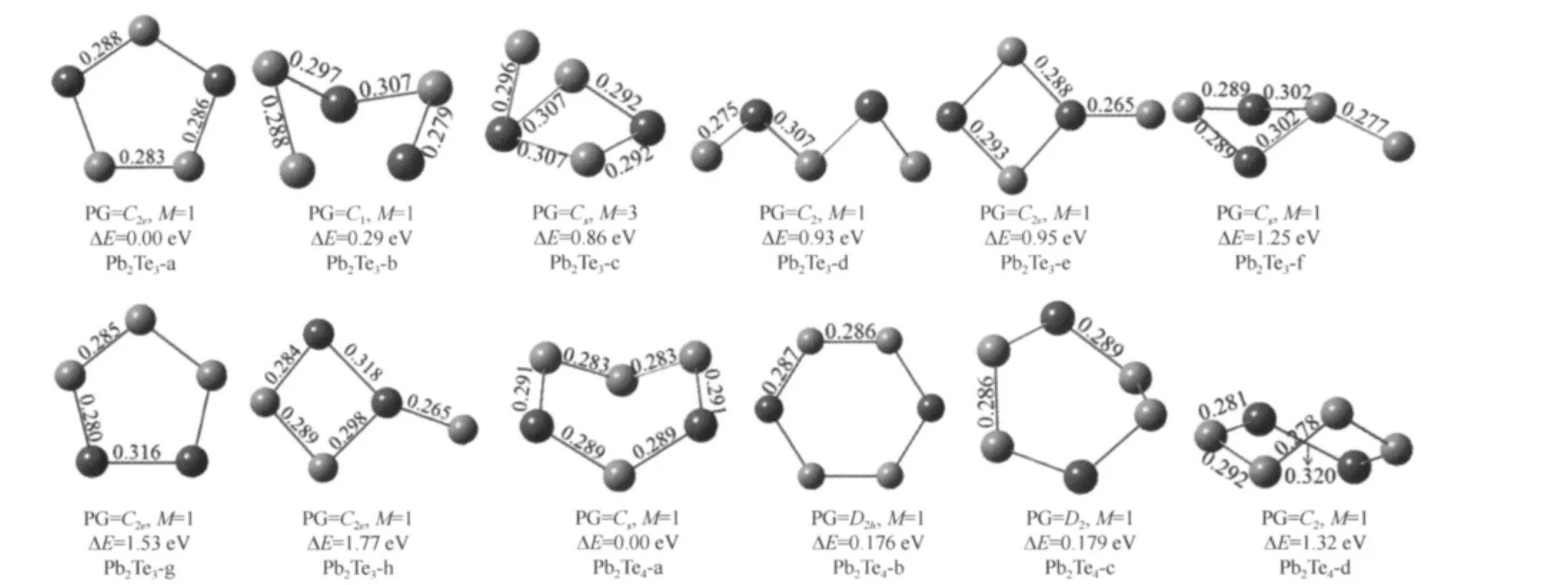
Fig.5 Stable geometries of Pb2Ten(n=3-4)clusters
2.2 Average binding energy per atom
The Ebof PbmTen(m+n≤6)clusters are given in Table 2.For Pb-rich clusters with one Te atom,the Ebincreases as the size of atoms increase,and the change trend of the Pb-rich clusters with two Te atoms is the same.On the contrary,for the Te-rich clusters with one Pb atom,the Ebdecreases with increasing the number of atoms,the Ebof Te-rich clusters with two Pb atoms decreases too.Compared the Ebof PbnTe with that of PbTen(n=2-5),the former are larger than the latter.The Ebof PbnTe2clusters are also larger than that of Pb2Tenclusters.It is shown that the Pb-rich clusters are more stable than Te-rich clusters.
2.3 Dissociation channels,dissociation energies and Mulliken charge populations
The stability of lead telluride clusters with different sizes and stoichiometries is required to illustrate the growth pattern of various nanostructures and to understand the pristine lead clusters. On the other hand,the study of the stability is helpful in finding the candidates of the building block of the cluster-assembled materials.In general,the cluster with large positive dissociation energy(ΔEd)has great stability[36].Therefore,in the following, we evaluate the ΔEdof the clusters.In our work,the possible dissociation channels have been carried out.The most probable ones with the corresponding ΔEdare presented in Table 3.
For the homonuclear Pbn(n=2-6)clusters,they prefer to dissociate one by one,the Pb atom is the popular fragment,while the homonuclear Ten(n=3-6)clusters are dissociated as dimers.In general,the dissociation energies of pure Pb clusters are larger than those of pure Te clusters,except the dimer.
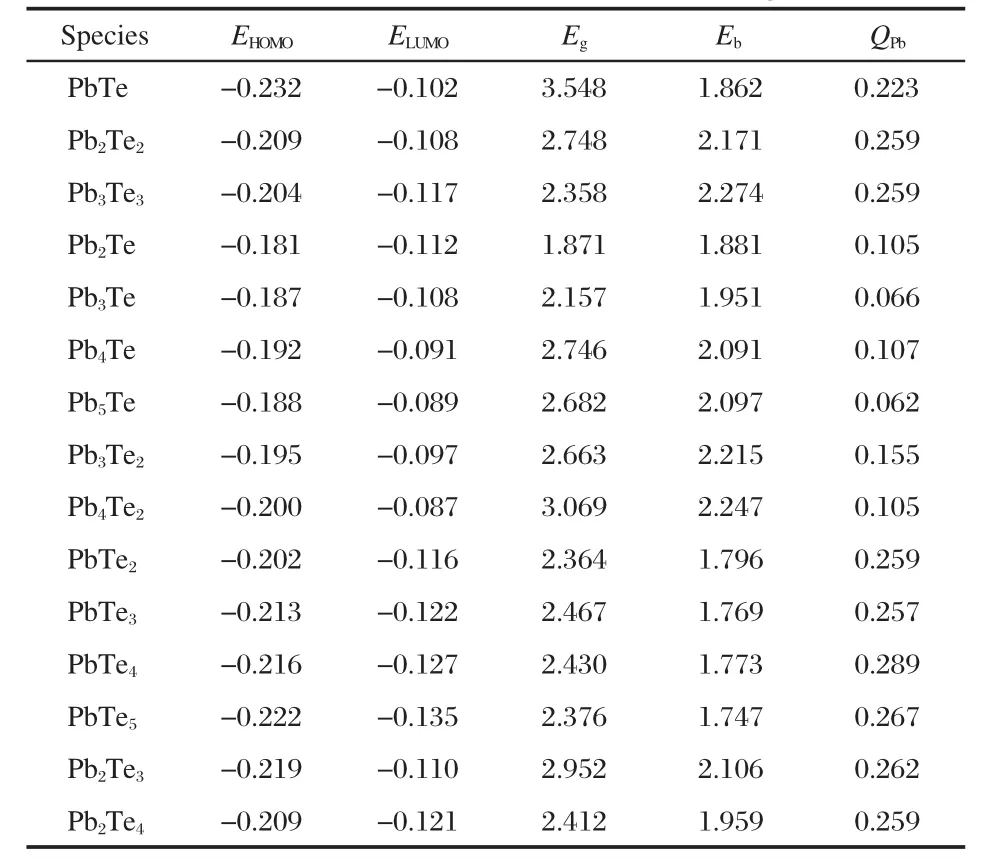
Table 2 Calculated HOMO,LUMO energies(in a.u.), HOMO-LUMO gap energies(Eg,in eV),Eb(in eV)for the most stable structures of the PbmTenclusters,and the the average Mulliken charge populations of Pb atoms(QPb,in e)
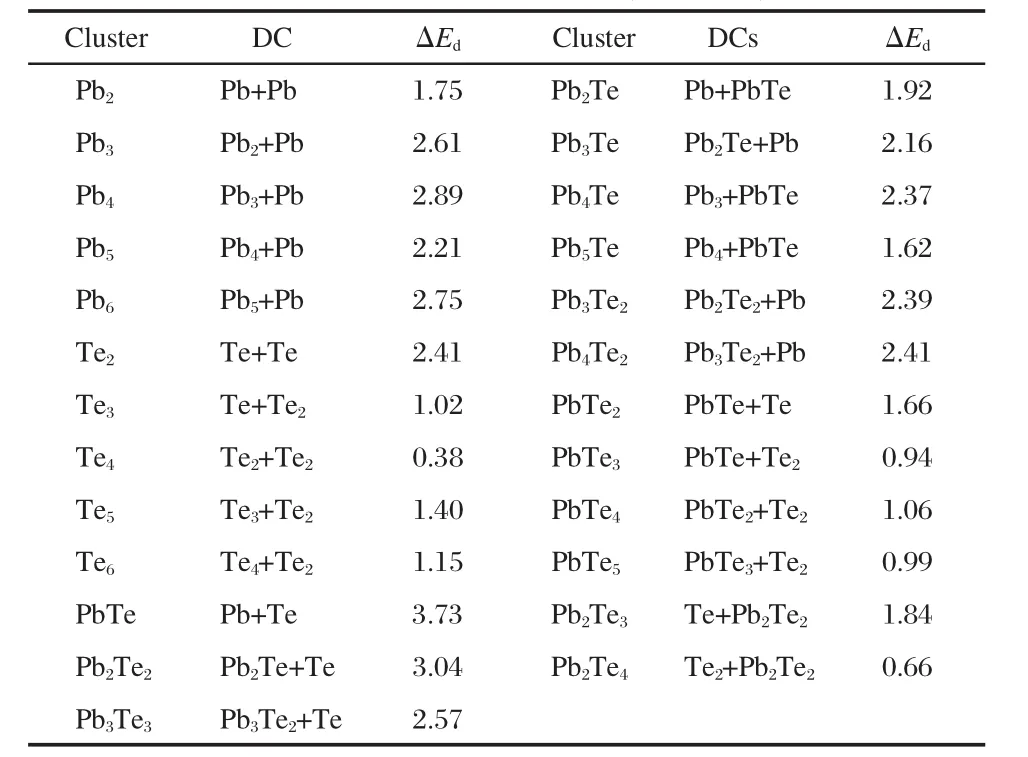
Table 3 The most favorable dissociation channels(DCs)and dissociation energies(ΔEd,in eV)for the most stable structures of the Pbn,Ten,PbmTen(m+n≤6)clusters
For the Pb-rich clusters,Pb atom prefers to dissociate first in many dissociation channels.However,for the dissociation channels of Pb4Te and Pb5Te clusters,the PbTe dimer is the favorable fragment.As for the most Te-rich clusters,it is found that Te2is the popular fragment,but for PbTe2and Pb2Te3clusters,the favorable dissociation products are PbTe and Pb2Te2,respectively. The(PbTe)n(n=1-3)clusters have the larger ΔEdamong the clusters with the same number of lead atoms.It is shown that the (PbTe)n(n=1-3)clusters have great stabilities,especially for the PbTe,the ΔEdis as large as 3.73 eV.Furthermore,PbTe and Pb2Te2clusters are generally the favorable dissociation fragments for most Pb-Te mixed clusters.The ΔEdof the PbnTe clusters(n=2-5)are larger than those of PbTen(n=2-5)clusters,and the ΔEdof PbnTe2(n=3-4)clusters are also larger than those of Pb2Ten(n=3-4)clusters.Therefore,the Pb-rich clusters(PbnTe, PbnTe2)are more stable than the corresponding Te-rich clusters (PbTen,Pb2Ten),this conclusion agrees well with the Ebanalysis.
In Table 2,the average Mulliken charge populations of Pb atoms are also presented,it can be seen that the Pb atoms exhibit positive charges,as electron donators.There is a degree of charge transfers from Pb to Te atoms in Pb-Te clusters due to the difference of electronegativities between them.According to Sanderson electronegativity[37],the value of Pb is 2.29,smaller than that of Te(2.62).The charge transfers can contribute to the stability of Pb-Te clusters.In the previous investigations,Chen et al.[38]found that the stability of Ag-core/Au-surface-segregated nanoalloys comes from a directional charge transfer induced by the icosahedral structural order,which is additional to that induced by the electronegativity difference between Au(2.4)and Ag(1.9)atoms.
2.4 HOMO-LUMO gap
The HOMO-LUMO gap can be used to characterize the stabilities and electronic properties of the clusters.The HOMOs, LUMOs and the HOMO-LUMO gaps of the mixed PbmTenclusters are listed in Table 2.It is obvious to discover that gaps of the studied clusters are in the range of 1.87-3.55 eV,which suggests that the mixed Pb-Te clusters present the semiconductorlike character.The PbTe cluster has the largest gaps(3.55 eV), also supporting its huge stability.Among clusters with the same number of lead atoms,the(PbTe)n(n=1-3)clusters have obvious large HOMO-LUMO gaps,it indicates that the(PbTe)nclusters have great stabilities,in agreement with the dissociation energies analysis.The 3D plots of HOMO and LUMO orbitals of (PbTe)nclusters are shown in Fig.6.As shown in Fig.6,the HOMO orbital of PbTe dimer presents π character derived from pzorbitals of Pb and Te atoms,as for the π*antibonding orbital (LUMO),it is mainly constructed with pzorbitals of Pb atom along with a small quantity of Te(pz).The bonding π HOMO orbital is responsible to the stability of PbTe dimer.In Pb2Te2cluster,the HOMO orbital presents π*character,which is completely originated from Pb 6p.The components of LUMO orbital are Pb(6p)and Te(5p).We can find the hybridization between two Pb 6p orbitals in the LUMO,however,the orbital overlap is not presented between Te and Pb atoms.The HOMO orbital of Pb3Te3cluster is completely derived from the 5p orbitals of three Te atoms,as for LUMO,the 6p atomic orbital of one limbic Pb mostly contributes the electron density.

Fig.6 3D plots of HOMO(H)and LUMO(L)of the(PbTe)n(n=1-3)clusters
3 Conclusions
In this work,the DFT with hybrid B3LYP functional and SDD basis set have been employed to investigate the geometrical structures and stabilities of small PbmTen(m+n≤6)clusters. Our conclusions can be summarized as follows.
(1)Based on the extensive search,the lowest energy structures and low-lying stable isomers have been found.
(2)The Eband ΔEdanalysis indicate that the Pbnand Pb-rich clusters are more stable than the Tenand Te-rich clusters,respectively.
(3)The HOMO-LUMO gaps of the studied PbmTenclusters are evidently moderate,in the range of 1.87-3.55 eV,suggesting the semiconductor-like behavior.
(4)The ΔEdanalysis indicates that the(PbTe)n(n=1-3)clusters have great stabilities,suggesting that they might be used as candidates of the building block of cluster-assembled materials,and that the PbTe cluster possesses the stronger stabilities relative to the other mixed PbmTenclusters.
1 Bailey,P.T.Phys.Rev.,1968,170:723
2 Martinez,G.;Schluter,M.;Cohen,M.L.Phys.Rev.B,1975,11: 651
3 Tung,Y.W.;Cohen,M.L.Phys.Rev.,1969,180:823
4 Mitchell,D.L.;Wallis,R.F.Phys.Rev.,1966,151:581
5 Rabii,S.Phys.Rev.,1968,167:801
6 Wei,S.H.;Zunger,A.Phys.Rev.B,1997,55:13605
7 Preier,H.Appl.Phys.,1979,20:189
8 Weinberg,I.J.Chem.Phys.,1963,39:492
9 Weinberg,I.J.Chem.Phys.,1962,36:1112
10 Baleva,M.;Mateeva,E.Phys.Rev.B,1994,50:8893
11 Nabi,Z.;Abbar,B.;Méçabih,S.;Khalfı,A.;Amrane,N.Comp. Mater.Sci.,2000,18:127
12 Hevine,Z.H.;Allan,D.C.Phys.Rev.Lett.,1989,63:1719
13 Lach-hab,M.;Papaconstantopoulos,D.A.;Mehl,M.J.J.Phys. Chem.Solids,2002,63:833
14 Grabecki,G.;Wróbel,J.;Zagrajek,P.;Fronc,K.;Aleszkiewicz, M.;Dietl,T.;Papis,E.;Kamińska,E.;Piotrowska,A.;Springholz, G.;Bauer,G.Physica E,2006,35:332
15 Mazzone,A.M.Comp.Mater.Sci.,2000,18:185
16 Rajesh,C.;Majumder,C.;Rajan,M.G.R.;Kulshreshtha,S.K. Phys.Rev.B,2005,72:235411
17 Wang,B.L.;Zhao,J.J.;Chen,X.S.;Shi,D.N.;Wang,G.H.Phys. Rev.A,2005,71:033201
18 Gingerich,K.A.;Cocke,D.L.;Miller,F.J.Chem.Phys.,1976, 64:4027
19 Dai,D.;Balasubramanian,K.Chem.Phys.Lett.,1997,271:118
20 Liu,H.T.;Wang,S.Y.;Zhou,G.;Wu,J.;Duan,W.H.J.Chem. Phys.,2007,126:134705
21 Xing,H.Z.;Xu,S.L.;Ding,Z.L.;Huang,Y.;Chen,X.S.;Wang, J.Q.;Shi,Y.Phys.Lett.A,2008,372:4694
22 Goddard,J.D.;Chen,X.Q.;Orlova,G.J.Phys.Chem.A,1999, 103:4078
23 Balasubramanian,K.;Dai,D.J.Chem.Phys.,1993,99:5239
24 Hassanzadeh,P.;Thompson,C.;Andrews,L.J.Phys.Chem., 1992,96:8246
25 Pan,B.C.Phys.Rev.B,2002,65:085407
26 Zhao,G.F.;Sun,J.M.;Liu,X.;Guo,L.J.;Luo,Y.H.J.Mol. Struct.-Theochem,2008,851:348
27 Ma,W.J.;Wu,H.S.Acta Phys.-Chim.Sin.,2004,20:290 [马文瑾,武海顺.物理化学学报,2004,20:290]
28 Hohenberg,P.;Kohn,W.Phys.Rev.,1964,136:B864
29 Kohn,W.;Sham,L.J.Phys.Rev.,1965,140:A1133
30 Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03. Revision B.02.Pittsburgh,PA:Gaussian Inc.,2003
31 Becke,A.D.J.Chem.Phys.,1993,98:5648
32 Lee,C.;Yang,W.T.;Parr,R.G.Phys.Rev.B,1988,37:785
33 Huber,K.P.;Herzberg,G.Molecular spectra and molecular structure IV.Constants of diatomic molecules.New York:Van Nostrand Reinhold Company Press,1979:530-531
34 Peköz,R.;Erkoç,S,.Physica E,2008,40:2921
35 Dai,D.;Balasubramanian,K.J.Chem.Phys.,1992,96:8345
36 Lei,X.L.;Zhu,H.J.;Wang,X.M.;Luo,Y.H.Acta Phys.-Chim. Sin.,2008,24:1655 [雷雪玲,祝恒江,王先明,罗有华.物理化学学报,2008,24:1655]
37 Sanderson,R.T.J.Chem.Educ.,1988,65:112
38 Chen,F.Y.;Johnston,R.L.Acta Materialia,2008,56:2374
猜你喜欢
北京航空航天大学学报(2021年9期)2021-11-02
北京航空航天大学学报(2021年7期)2021-08-13
北京航空航天大学学报(2021年6期)2021-07-20
泰山学院学报(2019年6期)2020-01-14
山西教育·招考(2019年12期)2019-09-10
物理化学学报(2018年6期)2018-03-08
物理化学学报(2015年5期)2015-02-28
天津商务职业学院学报(2015年2期)2015-02-28
天津商务职业学院学报(2015年1期)2015-02-28
物理化学学报(2013年4期)2013-07-25
