
取代基对甲酰胺二聚体中N-H…O=C氢键强度的影响
2013-01-10 03:38郑艳萍王敬微
通化师范学院学报 2013年8期
郑艳萍,王敬微
(通化师范学院 化学学院,吉林 通化 134002)
氢键是最重要的相互作用之一,它影响着从无机到生物的许多体系,决定着分子构造、分子聚集状态和功能[1-2].对氢键强度和本质进行深入研究具有重要意义[3-8].在本文中,我们利用二级微扰理论MP2方法,选取一系列N-H…O=C氢键二聚体为研究对象,深入探讨不同取代基对N-H…O=C氢键强度的影响.
1 计算方法
图1为本文研究的5个氢键二聚体.使用MP2/6-31+G**方法优化几何结构,并进行频率计算来确定所得的结构是稳定构型.在MP2/6-31+G**几何结构基础上,使用MP2/6-311++G(d,p)、MP2/6-311++G(3df,2p)和MP2/aug-cc-PVTZ方法计算了单点能并进行了基组重叠误差(BSSE)校正[9].全部计算采用Gaussian03程序包完成.
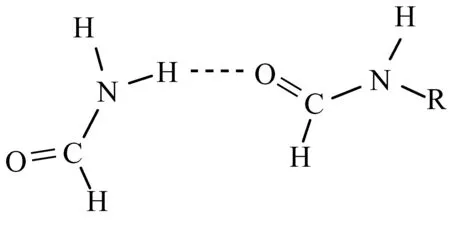
图1 5个氢键二聚体体系(R=-Me,-NH2,-H,-CONH2,-CF3)
2 结果与讨论
2.1 几何构型
图2给出了本文研究的5个氢键二聚体的优化结构及相应的几何参数.由图2可以看出,与甲酰胺二聚体(R=-H)相比,氢键受体分子中的供电子基团使氢键键长r(H…O)缩短,使N-H键长r(N-H)和C=O键长r(C=O)伸长,吸电子基团使氢键键长r(H…O)伸长,使N-H键长r(N-H)和C=O键长r(C=O)缩短.例如:甲酰胺二聚体(R=-H)的r(H…O)为1.973Å,r(N-H)为1.014Å,r(C=O)为1.235Å.当取代基为供电子基团-Me时,r(H…O)为1.952Å,缩短了0.021Å,r(N-H)与r(C=O)为1.015Å和1.239Å,分别伸长了0.001Å和0.004Å.取代基为供电子基团-NH2时,r(H…O)为1.962Å,缩短了0.011Å,r(N-H)与r(C=O)为1.014Å和1.236Å,r(C=O)伸长了0.001Å.当取代基为吸电子基团-CONH2时,r(H…O)为2.013Å,伸长了0.040Å,r(N-H)与r(C=O)为1.012Å和1.231Å,分别缩短了0.002Å和0.004Å.取代基为吸电子基团-CF3时,r(H…O)为2.043Å,伸长了0.07Å,r(N-H)与r(C=O)为1.011Å和1.227Å,分别缩短了0.003Å和0.008Å.由图2还可看出,不论取代基为供电子基团还是吸电子基团,二聚体中N-H键长r(N-H)和C=O键长r(C=O)与单体中的r(N-H)和r(C=O)比较都有所伸长.例如:当取代基为供电子基团-Me时,r(N-H)为1.015Å,r(C=O)为1.239Å,这与单体中的1.005Å和1.232Å比较,分别伸长了0.01Å和0.007Å.当取代基为吸电子基团-CF3时,r(N-H)为1.011Å,r(C=O)为1.227Å,这与单体中的1.005Å和1.221Å比较,都伸长了0.006Å,表明二聚体中氢键的形成使得N-H和C=O键变弱.
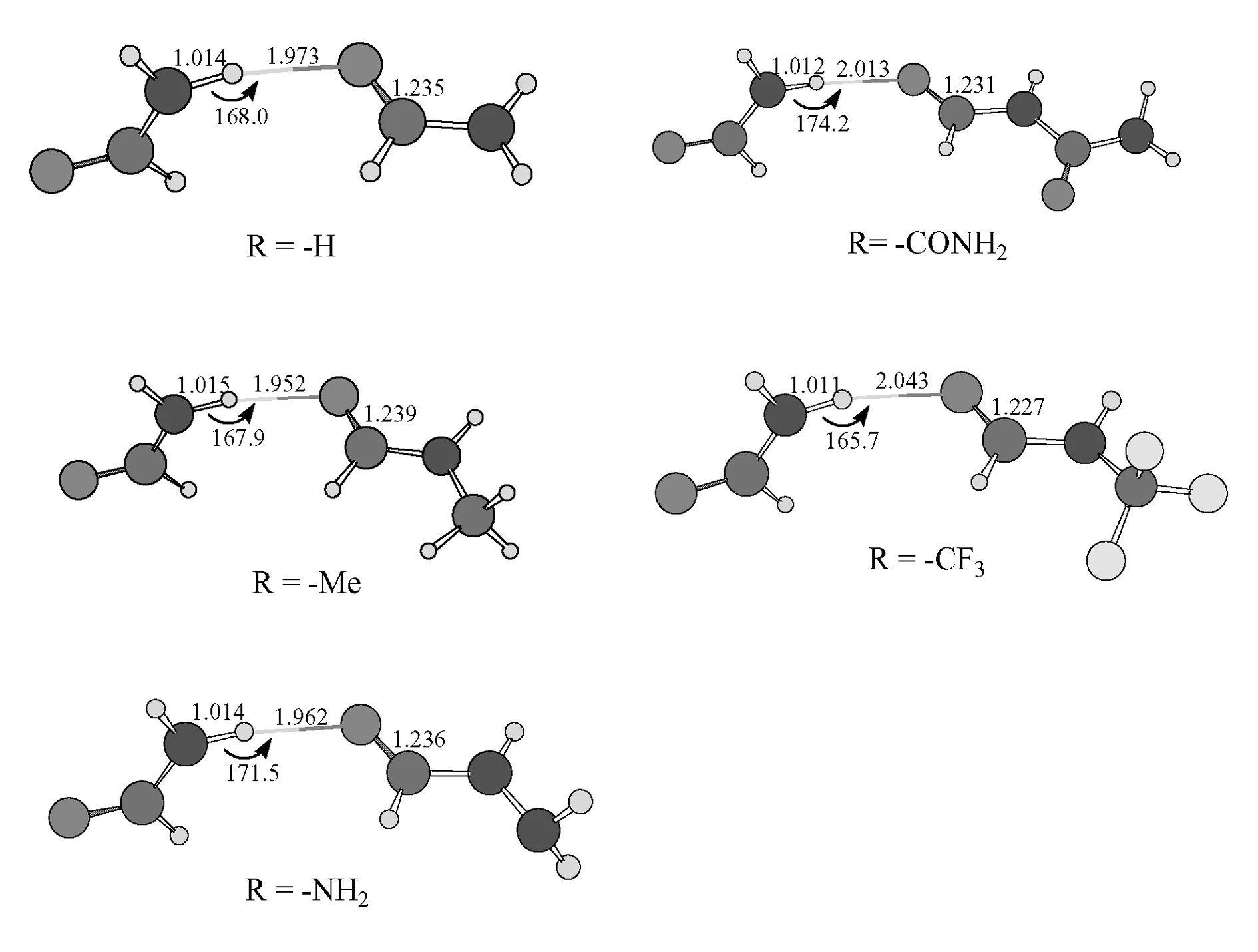
图2 MP2/6-31+G**水平优化的5个N-H…O=C氢键体系的几何结构
2.2 氢键能量
表1中第2~3列,4~5列,6~7列,8~9列给出了在MP2方法下分别使用6-31+G**、6-311++G(d,p)、6-311++G(3df,2p)和aug-cc-PVTZ基组计算得到的5个体系的氢键能量.
由表1可以看出,MP2/6-31+G**(BSSE)计算得到的甲酰胺二聚体(R=-H)的氢键能量为-6.04kcal/mol,MP2/6-311++G(d,p)(BSSE)的结果为-5.99kcal/mol,MP2/6-311++G(3df,2p)(BSSE)的结果为-6.80kcal/mol,MP2/aug-cc-PVTZ(BSSE)的结果为-6.97kcal/mol.我们使用MP2/aug-cc-PVTZ(BSSE)的计算结果(-6.97kcal/mol)与文献报道的甲酰胺二聚体中N-H…O=C的平均氢键能量-7.1kcal/mol[4]相符.与MP2/aug-cc-PVTZ计算结果比较,MP2/6-31+G**方法与MP2/6-311++G(d,p)方法计算的结果与之分别相差了0.93kcal/mol和0.98kcal/mol,而MP2/6-311++G(3df,2p)方法计算结果与之仅相差0.17kcal/mol,说明使用MP2/6-311++G(3df,2p)方法可以得到比较可靠的结果.由表1可以看出,MP2/aug-cc-PVTZ方法下不考虑BSSE校正时高估氢键相互作用能大约0.6~0.7kcal/mol.由表1还可以看出,用不同的基团取代质子受体分子中的氢原子对N-H…O=C氢键强度的影响是不同的.例如:MP2/aug-cc-PVTZ计算结果表明,甲酰胺二聚体(R=-H)的氢键相互作用能为-6.97kcal/mol.当取代基为供电子基团-Me时,二聚体的氢键相互作用能为-7.55kcal/mol,增强了0.68kcal/mol.取代基为供电子基团-NH2时,二聚体的氢键相互作用能为-7.16kcal/mol,增强了0.19kcal/mol.当取代基为吸电子基团-CONH2时,二聚体的氢键相互作用能为-5.74kcal/mol,减弱了1.23kcal/mol.取代基为吸电子基团-CF3时,二聚体的氢键相互作用能为-5.06kcal/mol,减弱了1.91kcal/mol.由此得出,供电子基团取代质子受体分子中的氢原子使氢键作用增强,而吸电子基团使氢键作用减弱.
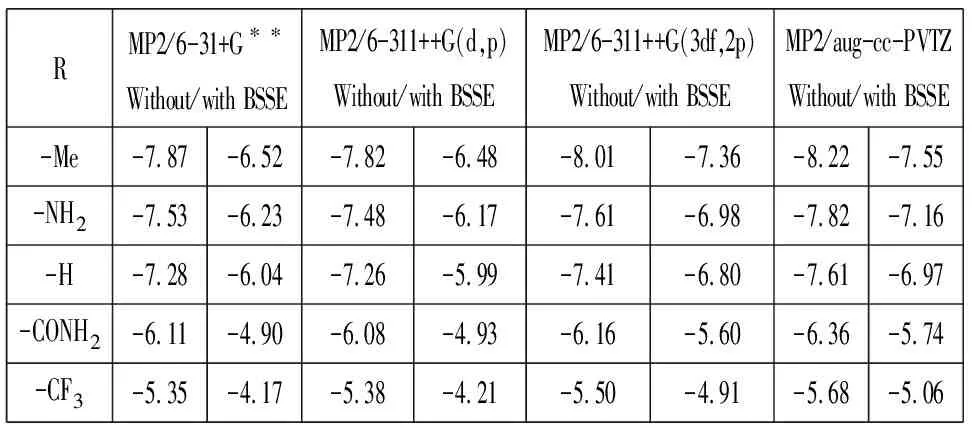
表1 MP2方法计算得到的5个N-H…O=C体系氢键能量 (kcal/mol)
2.3 自然键轨道(NBO)分析
表2给出了本文研究的5个氢键二聚体体系的自然键轨道(NBO)分析计算结果,其中qH、qO分别为参与形成氢键的氢原子和氧原子的电荷,ΔqH为二聚体中参与形成氢键的qH与单体分子中相应的氢原子电荷的差值,ΔqO为二聚体中参与形成氢键的qO与单体分子中相应的氧原子电荷的差值.qD、qA分别为质子供体分子和质子受体分子的电荷,Eij为N-H…O=C氢键中氧原子的孤对电子(n)对N-H反键轨道σ*的二阶稳定化能,EHB为使用MP2/aug-cc-PVTZ(BSSE)方法计算得到的氢键能量.
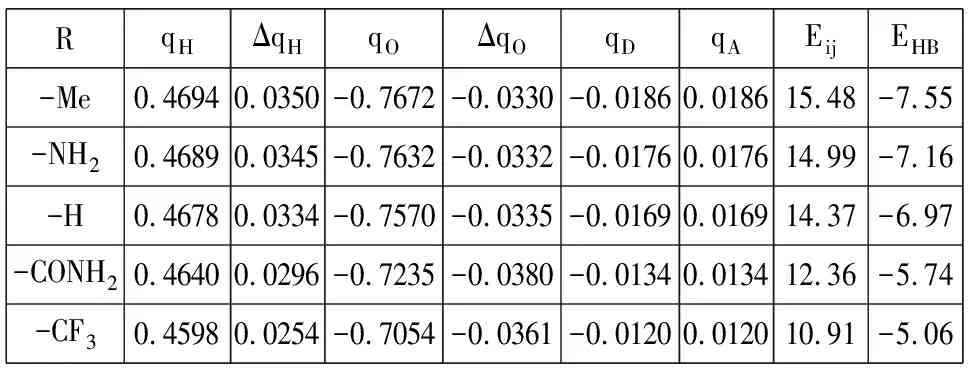
表2 MP2/6-31+G**水平5个氢键体系的自然键轨道分析结果及氢键能量(kcal/mol)
由表2第2~3列可以看出,与甲酰胺二聚体(R=-H)中参与形成氢键的氢原子电荷0.4678比较,当取代基为供电子基团-Me时,qH为0.4694,正电荷增加了0.0016.取代基为吸电子基团-CF3时,qH为0.4598,正电荷减少了0.008.由表2中第4~5列可以看出,与甲酰胺二聚体(R=-H)中参与形成氢键的氧原子电荷-0.7570比较,当取代基为供电子基团-Me时,qO为-0.7672,负电荷增加了0.0102,这是由于-Me具有供电子能力,从而使参与形成氢键的氧原子电荷增加;取代基为吸电子基团-CF3时,qO为-0.7054,负电荷减少了0.0516,这是由于-CF3具有吸电子能力,使参与形成氢键的氧原子电荷降低.对于其它的基团都有类似的结果,即氢键强度越强,氢键N-H…O=C中氢原子的电荷越正,氧原子的电荷越负.由表2中第6~7列可以看出,与甲酰胺二聚体(R=-H)单体分子间电荷转移量0.0169比较,当取代基为供电子基团-Me时,单体分子间电荷转移量为0.0186,电荷转移量增多;当取代基为吸电子基团-CF3时,单体分子间电荷转移量为0.0120,电荷转移量减少.表2中第6、第7和第9列数据表明,单体分子间电荷转移的越多,氢键相互作用越强.
氢键作用相对强弱还可以用质子受体的孤对电子对(n)和质子供体的反键σ*轨道之间n→σ*的相互作用来描述.由表2可以看出,与甲酰胺二聚体的二阶稳定化能Eij比较,用不同的取代基取代质子受体分子中的氢原子,供电子基团使得n(O)→σ*(N-H)的二阶稳定化能Eij增加,而吸电子基团使得n(O)→σ*(N-H)的二阶稳定化能Eij减弱.例如:甲酰胺二聚体(R=-H)的二阶稳定化能Eij为14.37kcal/mol,取代基为供电子基团-Me时,二阶稳定化能Eij为15.48kcal/mol,增加了1.11kcal/mol.取代基为吸电子基团-CF3时,二阶稳定化能Eij为10.91kcal/mol,减弱了3.46kcal/mol.表2中第8~9列数据表明,氢键强度越强,二阶稳定化能越大.
3 结论
本文研究表明,可以通过改变取代基来影响二聚体中N-H…O=C氢键的强度.取代基为供电子基团时,氢键强度增强,取代基为吸电子基团时,氢键强度减弱.并且氢键强度越强,N-H…O=C氢键中氢原子电荷越正,氧原子电荷越负,单体分子间电荷转移越多,质子受体的孤对电子对(n)和质子供体的反键轨道σ*之间n(O)→σ*(N-H)的二阶稳定化能越大.
参考文献:
[1]Zhao G.J,Liu J.Y,Zhou L.C,et al.Site-selective photonduced electron transfer from alcoholic solvents to the chromophore facilitated by hydrogen bonding:A new fluorescence quenching mechanism[J].J.Phys.Chem.B,2007,111:8940.
[2]Huang Z M,Han K,Li H P,et al. Hydrogen bonding effects on the nonlinear optical properties of 2-(N-methyl) amino-5-nitropyridine molecule [J].J.At.Mol.Phys.,2007,24:518.
[3]Li X,Liu L,Schlegel H B.On the physical origin of blue-shifted hydrogen bonds[J].J.Am.Chem.Soc.,2002,124:9639-9647.
[4]Vargas R,Garza J,Friesner R.et al.Strength of N-H…O=C and C-H…O=C bonds in formamide and N-methylacetamide dimer[J].J.Phys.Chem.A.,2001,105:4963-4968.
[5]Wang C S,Zhang Y,Gao K,et al.A new scheme for determining the intramolecular seven-membered ring N-H…O=C hydrogen-bonding energies of glycine and alanine peptides[J].J.Chem.Phys.,2005,123:4307.
[6]Sun C L,Wang C S.Theoretical studies on the binding energy of β-sheet models[J].Sci.China.Ser. B-Chem.,2009(12):223-228.
[7]Tan H W,Qu W W,Chen G J.The role of charge transfer in the hydrogen bond cooperative effect of cis-N-methylformamide oligomers[J].J.Phys.Chem.A.,2005,109:6303-6308.
[8]Zhang Y,Wang C S.Estimation on the intramolecular 10-membered ring N-H…O=C hydrogen-bonding energies in glycine and alanine peptides[J].J.Comput.Chem.,2009,30:1251.
[9]Boys S F,Bermardi F.The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors[J].Mol.Phys.,1970,19:553.
猜你喜欢
波谱学杂志(2021年3期)2021-09-07
——以高中化学“氢键”的教学为例
教学月刊(中学版)(2020年13期)2020-12-29
青岛大学学报(工程技术版)(2019年2期)2019-09-10
农药科学与管理(2019年10期)2019-04-20
天津师范大学学报(自然科学版)(2016年4期)2016-12-14
中国塑料(2016年11期)2016-04-16
中国粮油学报(2016年1期)2016-02-06
中学化学(2015年12期)2016-01-19
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
