
自身炎症性疾病诊治
2013-04-09 01:44沈敏
中华临床免疫和变态反应杂志 2013年3期
沈 敏
(中国医学科学院 北京协和医学院 北京协和医院风湿免疫科,北京 100730)
自身炎症性疾病(autoinflammatory diseases,AUID)是近几年被逐渐认识的一组疾病,由于基因突变使编码蛋白发生改变,导致固有免疫失调而引起全身性炎性反应。
固有免疫系统由某些白细胞(中性粒细胞、树突细胞、巨噬细胞、自然杀伤细胞)、前炎性信号蛋白(细胞因子)及补体系统组成。白介素(interleukin,IL)-1、IL-6和肿瘤坏死因子α(tumor necrosis factor-α,TNF-α)是固有免疫系统中最常见且重要的前炎性细胞因子。所有与AUID相关的基因突变都可致固有免疫炎性信号失调,引起前炎性状态改变,最终导致炎性体活化。炎性体是一种细胞浆多蛋白复合物,在前炎性因子如IL-1β的产生和释放中起到了关键性作用[1],并在各种不同AUID的发病机制中发挥作用。多种信号参与了炎性体的形成,如微生物、内源性产物如胆固醇和尿酸、前炎性细胞因子和趋化因子。
尽管临床医师已经提高了对AUID的认识,但是由于其临床表现多样,常出现误诊或延迟诊断。AUID由于其遗传性的特点,大多发病较早,从出生后数小时到十多岁青少年期均可发病,少数患者成年后发病。AUID临床表现为复发性全身性炎性反应,绝大多数患者表现为突发周期性发热,伴急相反应物升高、皮疹、浆膜炎、淋巴结肿大和关节炎等。而在无症状的发作间期,患者身体状况和生长发育同正常人,急相反应物也完全正常。
AUID包括4组疾病,即周期性发热、cryopyrin相关周期性综合征(cryopyrin-associated periodic syndromes,CAPS)、肉芽肿性疾病及化脓性疾病[2]。以下对各种AUID的发病机制、临床表现和治疗新进展进行综述。
周期性发热
该类型AUID主要包括3种单基因疾病:家族性地中海热(familial mediterranean fever,FMF),甲羟戊酸激酶缺乏症(mevalonate-kinase deficiency,MKD),肿瘤坏死因子受体相关周期性综合征(tumour necrosis factor receptor-associated periodic syndrome,TRAPS)。
家族性地中海热
FMF是遗传性复发性炎性疾病中最常见的一种,属常染色体隐性遗传病,最早报道于20世纪初,主要发病人群是地中海裔[3]。
发病机制:FMF相关基因定位于16号染色体短臂,称作地中海热基因(MEditerranean FeVer,MEFV)。MEFV突变造成其编码蛋白pyrin失调,进一步使IL-1产生增加。蛋白pyrin的几个重要结构域与AUID发病相关:(1)位于N端的pyrin结构域(Pyd);(2)位于C端的B30.2或SPRY结构域。Pyrin通过同型Pyd与调亡斑点蛋白(apoptotic speck protein,ASC)相互作用,ASC同时介导了NF-κB和前caspase-1活化,继而促进IL-1β的产生和释放,最终导致细胞调亡[4]。另外,早期研究表明pyrin通过Pyd和B30.2结构域共同与炎性体作用。近来在动物模型中发现,一种与化脓性无菌性关节炎-坏疽性脓皮病-痤疮(pyogenic sterile arthritis, pyoderma gangrenosum and acne,PAPA)综合征相关的蛋白PSTPIP1也能激活pyrin,导致Pyd与ASC相互作用[5]。
临床表现:FMF通常发病年龄为5~15岁,约20%患者20岁以后发病。发热持续<72 h,可自行好转,复发周期无规律。发热时常伴急性浆膜炎,腹膜炎和单侧胸膜炎发生率分别达90%和40%以上。最常见的症状为剧烈腹痛和胸痛,腹痛程度严重似急性阑尾炎以致行剖腹手术。少数患者可出现阴囊炎和心包炎。约70%的患者可出现大关节(主要是膝、髋和踝)关节痛或关节炎。最常见的皮肤改变为下肢丹毒样红斑,发生率约40%,病理检查常见弥漫性、中性粒细胞为主的炎细胞浸润。还有报道FMF可出现血管炎,如过敏性紫癜、结节性多动脉炎和贝赫切特综合征。FMF的诱发因素包括应激、病毒感染或药物。FMF发作的前驱症状包括潜在炎性反应受累部位不适,或者各种生理性、情绪和躯体性主诉,例如易激惹、头晕、食欲增长和味觉改变。
淀粉样变是FMF最主要的并发症,肾脏淀粉样变则为FMF患者最主要的死因。FMF淀粉样变主要发生于较早发生严重炎性反应的患者(1型FMF)或发作间期急性反应物仍高的患者,偶可见于临床无显著炎性反应发作者(2型FMF)[6]。基因突变和(或)环境因素均参与了淀粉样变的发生。其他慢性表现,如包裹性腹膜炎和累及髋、膝的慢性毁损性关节炎、脾脏肿大都非常少见。
诊断:MEFV基因分析有时很难正确解读。不同等位基因上2个基因突变有助于确诊,但若只有1个MEFV基因突变,而患者具备FMF的临床表现,仍然不能除外FMF。此外,尽管有5种突变最常见(占所有突变的85%),但也存在一些罕见或未知的突变。因此诊断时需结合临床表现、基因检测和对秋水仙碱治疗有效综合评价。
治疗:每日口服秋水仙碱是FMF最主要的治疗方法,近70%的患者症状可完全消失,约25%的患者炎性发作次数减少、淀粉样变减轻。秋水仙碱常用剂量是1 mgd,无效者可以每3~6个月增加1次剂量,每次增加0.5 mg,总量不超过2.5 mgd。α干扰素无确实疗效。近期有报道抗IL-1拮抗剂如anakinra(阿那白滞素)和canakinumab(人抗IL-1β单克隆抗体)有效。
甲羟戊酸激酶缺乏症
MKD最早报道于1984年,高IgD曾被认为是诊断此病的血清标志物,故又称高IgD综合征。但后来发现位于染色体12q24上的甲羟戊酸激酶(mevalonate kinase,MVK)基因突变才是真正病因[7],且IgD升高的敏感性和特异性都很低,所以现在不再称高IgD综合征,而代之以MKD。MKD发病人群除北欧外还包括地中海盆地和亚洲。
发病机制:异戊二烯生物合成途径中的关键酶包括羟甲基戊二酰辅酶A还原酶与MVK。MVK基因突变致蛋白保守区域突变(多为无意义突变),使MVK活性下降(仅为正常值1~5%)[8],直接导致异戊二烯终末产物缺乏(主要为牛儿基焦磷酸),后者致caspase-1活性增加,IL-1β过度分泌,触发炎性反应[9]。
临床表现及诊断:MKD最常发生于婴儿,70%患者2岁以内发病,几乎所有患者都在10岁以内起病,也有成人发病的报道。注射疫苗可能诱发本病。突发发热常持续4~6 d。90%患者出现颈部淋巴结肿大,80%伴皮疹(掌跖红斑),约70%伴严重腹痛、呕吐、腹泻、头痛等,少见口腔浅表溃疡。大多数患者有关节痛或非对称性寡关节炎[10]。随着年龄增长MKD病情严重程度下降,但部分患者可持续到成年。淀粉样变罕见,发热时急相反应物升高。发作期和缓解期IgD水平升高(>100 Uml),但特异性很低,也可见于Still病、FMF或吸烟者。发热时尿甲羟戊酸排泄增多以及MVK活性降低,特异性和敏感性更高[11],但需要高度专业的实验室技术,所以很难用来作为筛选试验。
MKD为常染色体隐性遗传疾病,MVK基因检测是诊断金标准。MKD临床表型轻重不一,与其基因表型不同有关。部分变异(V310M,A334T)导致严重细胞MVK活性受损,出现一种特殊综合征——甲羟丙二酸尿症,主要表现为智力发育迟滞、共济失调、生长缓慢、肌病、白内障及反复发热;最常见的V377I变异导致MVK活性轻度下降,表现为轻度MKD;部分患者表现为中间型[12]。值得注意的是,约30%患者虽然MVK基因检测结果阴性,却仍然符合MKD临床诊断标准[13]。
肿瘤坏死因子受体相关周期性综合征
TRAPS是一种常染色体显性遗传病,因p55 TNF受体(或称TNFR1A)突变所致,该受体由TNF超家族受体1A基因(TNF super family receptor 1A gene,TNFRSF1A)编码。TRAPS最早报道见于北欧人群,因此又被称为家族性爱尔兰热,但也可见于美国黑人、日本人和地中海裔人。
发病机制:目前已发现TNFRSF1A的总共114个序列变异,其中75个与TRAPS表型相关,大多数属无意义突变。患者血浆TNF可溶性受体的量和质均存在异常。游离TNFRs从细胞膜脱落产生大量可溶性受体,后者能通过与膜结合受体竞争性结合而清除循环TNF。TNFRSF1A突变可能干预脱落过程,导致患者缺乏对TNF的适当抑制。此外,TNFR1可通过NF-κB触发细胞活化,或通过活化前调亡caspase来触发调亡,而突变TNFR1则诱导调亡缺陷,致炎细胞持续活化。且突变TNFR1与细胞膜结合存在缺陷,无法将细胞内质网转运到细胞膜,内质网潴留致线粒体反应氧自由基过度释放,成为炎性反应持续的又一可能机制。
临床表现及诊断:TRAPS患者发热期通常持续1~3周,比FMF和MKD更长。婴儿至成人各阶段均可发病。患者可出现明显腹痛,约半数患者曾因此进行手术[16]。大多数患者可出现各种皮疹,如荨麻疹及斑疹等。临床特点为肿胀、发热和触痛性红斑,红斑大小不等、边界不清,常累及四肢,也可出现在前胸,通常呈游走性。TRAPS发作期的另一显著表现是假性蜂窝织炎,常伴肌痛,还可出现胸痛、阴囊疼痛、关节炎、眶周水肿和结膜炎。发作期急相反应物升高,发作间期甚至也不能完全恢复正常。TNFRSF1A基因检测有助于诊断,有家族史患者常见,散发病例中阳性率不高[17]。
治疗:秋水仙碱对于预防TRAPS反复发生无效。激素可有效减低病情程度和病程时间。但因炎性反应持续,需每日服用激素,为避免出现激素依赖,常需加用其他抗炎药物。依那西普治疗TRAPS及肾脏淀粉样变有效,但亦有部分患者可能无效或有效性逐渐下降。其他TNF抑制剂效果不佳,甚至有报道,部分TRAPS患者应用英夫利西单抗后反而使炎性反应加重。近来,越来越多的证据表明,anakinra对控制临床症状具有更良好且持久的作用。
核苷酸寡聚化结构域样受体蛋白12相关AUID
此病最早报道于2个来自瓜德罗普岛的家系,与cryopyrin相关周期性综合征(cryopyrin-associated periodic syndromes,CAPS)相似,临床表现为反复发热和冷敏感,寒冷或疲乏后出现一过性荨麻疹、肌肉骨骼疼痛和全身不适[18],伴神经性耳聋、阿弗它溃疡、淋巴结肿大、腹痛和急相反应物升高。病因为核苷酸寡聚化结构域样受体(nucleotide-binding oligomerization domain like receptor,NLR)家族的另一成员——NLR蛋白12(NLR protein 12,NLRP12)基因无意义突变。NLRP12在前炎性NF-κB通路的调节中发挥了作用,而继发于氧化还原失调的IL-1加速释放可能是疾病的另一发病机制。
cryopyrin相关周期性综合征
CAPS包括家族性冷自身炎症综合征(familial cold autoinflammatory syndrome,FCAS),Muckle-Wells综合征(Muckle-Wells syndrome,MWS),慢性婴儿神经皮肤关节综合征(chronic infantile neurological cutaneous and articular syndrome,CINCA)。3种疾病均属于常染色体显性遗传性疾病。
发病机制
本病由单个基因NLRP3(又称冷诱发自身炎性综合征1,cold-induced autoinflammatory syndrome 1, CIAS 1)的不同突变所致,该基因编码蛋白cryopyrin。NLRP3基因突变可见于约70%CAPS患者。Cryopyrin是炎性体关键蛋白,其寡聚化并与适配体蛋白ASC结合,直接激活caspase-1,前IL-1β活化并释放,导致炎性反应发生。
临床表现
CAPS最典型的症状是反复发热、荨麻疹和中枢神经系统炎性反应。多于婴幼儿期发病,成人发病临床表现不典型。
FCAS主要表现为寒冷暴露诱发短期(通常24 h内)发热和荨麻疹。关节痛和结膜炎也较常见。其他症状包括寒冷诱发的满头大汗、睡意、头痛、异常口渴和恶心。
MWS主要表现为反复发生的荨麻疹和发热,与冷暴露关系不密切。还可出现头痛(无菌性脑膜炎)、感音神经性聋和多关节炎。发热期急相反应物升高,发热间期仍可持续轻度升高。淀粉样变是该病晚期并发症。
CINCA是cyopyrin基因突变中最严重的类型。在出生后头几周即可出现荨麻疹样皮疹。患者多表现为前额突出、鞍鼻和面中部发育不良。骨骼受累最突出的特征是骨过度生长,主要累及膝(包括髌骨)和手足远端。慢性炎性多关节炎有时可导致骨侵蚀。中枢神经系统表现包括慢性无菌性脑膜炎、颅压升高、脑萎缩、脑室扩张、感音神经性聋、慢性视乳头水肿伴视神经萎缩和视力下降。智力发育迟滞和癫痫也有报道。患者急相反应物质、中性粒细胞持续升高,并有慢性贫血。
治疗
FCAS病情较轻,通过避免寒冷刺激常可使病情自然缓解。MWS与CINCA予抗IL-1治疗。Anakinra起始剂量为每日1 mgkg皮下注射。首次注射后症状好转,且1周之内完全缓解[19],治疗3年随访发现,所有CINCA患者症状控制良好,约30%患者听力下降可有效改善。其他IL-1拮抗剂,如rilonacept(列洛西普)和canakinumab(人抗IL-1β单克隆抗体)也同样有效[20-21]。
肉芽肿性疾病
Blau综合征属于AUID中的肉芽肿性疾病,也称家族性幼年性系统性肉芽肿,是常染色体显性遗传病,主要特点是非干酪样坏死性肉芽肿炎性反应,累及关节、皮肤和葡萄膜,出现关节炎、皮炎和葡萄膜炎三联征。病变基因NOD2CARD15属于NLR超家族,编码一种含NACHT域的蛋白。NOD2CARD15识别并激活胞壁酰二肽后,诱导NF-κB和IL-1β活化并释放[22]。
Blau综合征常于出生后几年内发病。显著腱鞘炎致沼泽样外观的对称性多关节炎最具特征。眼受累特征性表现为中间葡萄膜炎或全葡萄膜炎。50%眼部受累患者发生白内障,约13出现继发性青光眼。大约90%患者可出现典型的棕褐色鳞片状鱼鳞病皮疹。予患者口服激素和免疫抑制剂(甲氨蝶呤、环孢素A)疗效不一。近来有个例报道,英夫利西单抗和抗IL-1治疗有效。
化脓性疾病
化脓性疾病包括PAPA综合征,Majeed综合征,IL-1受体拮抗剂缺乏症(deficiency of the interleukin-1 receptor antagonist,DIRA)
化脓性无菌性关节炎-坏疽性脓皮病-痤疮
PAPA综合征是一种由于编码CD2结合蛋白1(CD2BP1)或PSTPIP1的基因突变所致疾病。常见临床表现为化脓性坏疽、囊性痤疮、化脓性无菌性关节炎[23]。幼儿常见寡关节炎,反复发作类似感染性关节炎,关节腔内大量无菌性、脓性、富含中性粒细胞的物质,最终导致显著滑膜和软骨破坏。皮肤表现通常发生于十几岁,特征性表现为周期性、复发性、进展性、溃疡性皮损,下肢多见,与脓性坏疽不易鉴别。也可见注射部位无菌性脓肿。PAPA综合征大多对口服激素有效。有个例报道应用抗TNF和抗IL-1治疗有效[24]。
Majeed综合征
本病以慢性复发性多灶性骨髓炎(chronic recurrent multifocal osteomyelitis,CRMO)、先天性红细胞生成性贫血和嗜中性粒细胞性皮肤病为特征,与位于染色体18p上的LPIN2基因突变相关[25]。与单纯CRMO不同的是,Majeed综合征患者骨骼表现发病早,发作频率和复发率高,缓解率低。先天性红细胞生成性贫血表现为外周血和骨髓小红细胞,需要反复输血。炎性皮病可表现为Sweet综合征或慢性脓疱病。也可出现反复发热和生长迟缓。非甾类抗炎药部分有效,短期口服激素能迅速控制症状,但可能激素依赖。秋水仙碱无效。脾切对纠正贫血可能有效。
IL-1受体拮抗剂缺乏症
DIRA是最近被发现的一种常染色体隐性遗传AUID。因IL1RN基因突变致IL-1受体拮抗剂缺乏、IL-1β活化释放。患儿出生后即发病,主要表现为多灶性骨髓炎、骨膜炎和脓疱病。皮肤表现为小脓疱聚集或泛发型脓疱病。骨骼表现包括溶骨性病变伴硬化性边缘、长骨远端和近端多发骨骺气球样变,肋骨和锁骨增宽、异位骨化或股骨干骺端近端骨膜隐形以及骨干骨膜抬高。急相反应物持续升高。anakinra治疗后病情戏剧性好转[26]。
多因素自身免疫性疾病
多因素AUID临床表现与遗传性AUID相似,也被认为主要与自身炎性反应相关,其中部分疾病也对IL-1拮抗剂显著有效。
1987年,Marshall等首次报道周期性发热-阿弗它口炎-咽炎-淋巴结炎(periodic fever, aphthous stomatitis, pharyngitis and adenitis,PFAPA),主要见于儿童,也可见于成人。尽管有家族性PFAPA报道[27],但未能证实其基因突变。近期研究发现,PFAPA患者体内存在异常T细胞调节的补体活化和IL-1产生[28]。PFAPA主要表现为规律性反复发热(通常精确到小时),持续5 d左右,间隔平均4周。其中咽炎最常见,与发热伴随出现,呈红斑或渗出性。口炎主要表现为发热前的浅小溃疡。发热时可出现颈部淋巴结肿大触痛,急相反应物升高。约60%患者可出现腹痛。发热间期上述症状完全正常。诊断主要依赖于临床。疾病呈现良性过程,可随年龄增长自发缓解。疾病发作早期给予单次剂量激素常可迅速缓解发热,但是有报道认为可能会缩短疾病发作间期。
痛风是一种由于单尿酸钠结晶沉积在关节和关节周围软组织造成疾病。有研究发现单尿酸钠结晶可激活炎性体,因此有学者认为痛风也属于一种多基因AUID。而事实证明IL-1阻滞剂可用于治疗秋水仙碱无效的痛风患者[29]。
全身型幼年特发性关节炎(systemic onset juvenile idiopathic arthritis,SoJIA)与遗传性AUID存在许多相同的临床表现,如全身性炎性反应、关节炎、皮疹、急相反应物升高。有研究发现,SoJIA患者体内存在普遍IL-1β信号,较多患者(40%至87%不等)对抗IL-1阻滞剂疗效显著[30]。
特发性复发性心包炎也在很多方面与AUID相似,anakinra疗效显著。
近来有证据表明,与AUID的固有免疫活化相同的发病机制在其他几种常见多因素疾病中也发挥了重要作用,例如II型糖尿病[31]和动脉粥样硬化[32]。
拟诊自身免疫性疾病的诊断评估
随着对AUID发病机制的不断深入了解,其诊断逐渐成为临床医师的巨大挑战。详实准确的病史采集和查体仍然是诊断关键。与感染、肿瘤及其他风湿免疫病的鉴别诊断亦很重要。监测急相反应物的变化有助于诊断。对于皮肤、骨骼、中枢神经系统、肾脏等特殊部位的针对性评估有助于AUID的分类诊断。对怀疑AUID的患者应进行基因分析,但需注意,约30%临床表现为AUID的患者最终基因分析结果为阴性。此外,基因分析费时费钱,并且需要较高实验室技术,因此并不能作为AUID常规筛查和确诊手段。短期给予秋水仙碱、激素或抗IL-1阻滞剂可作为诊断性治疗方法[33](图1)。
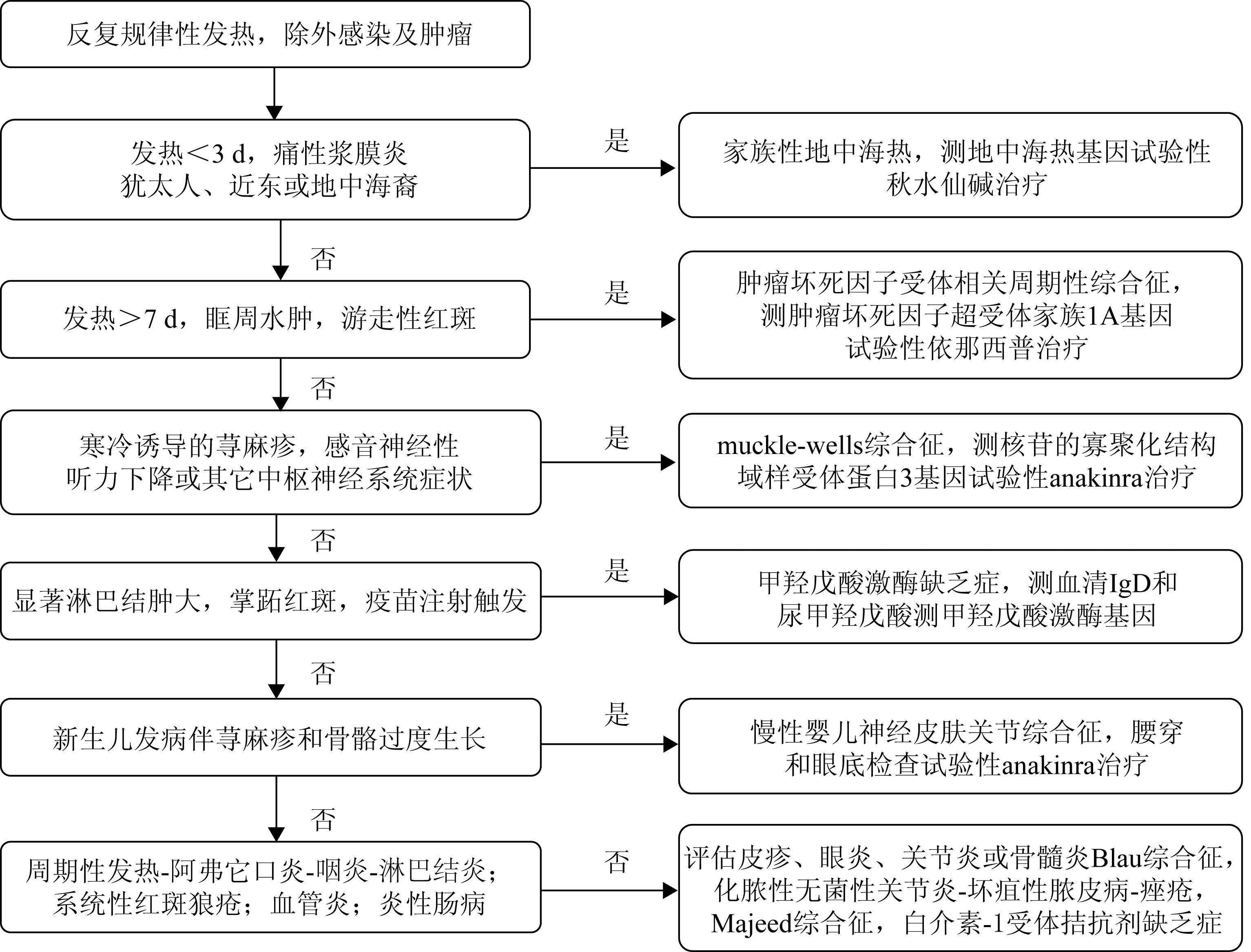
图1 自身炎性疾病诊断流程Fig 1 Diagnostic algorithm of autoinflammatory diseases
[1]Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta[J]. Mol Cell, 2002, 10:417-426.
[2]Federici S, Caorsi R, Gattorno M. The autoinflammatory diseases[J]. Swiss Med Wkly, 2012, 142:w13602.
[3]Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world[J]. Arthritis Rheum, 2009, 61:1447-1453.
[4]Chae JJ, Wood G, Richard K, et al. The familial Mediterranean fever protein, pyrin, is cleaved by caspase-1and activates NF-kappa B through its N-terminal fragment[J]. Blood, 2008, 112:1794-1803.
[5]Chae JJ, Cho YH, Lee GS, et al. Gain-of-function pyrin mutations induce NLRP3 protein-independent interleukin1beta activation and severe autoinflammation in mice[J]. Immunity, 2011, 34:755-768.
[6]Ben-Zvi I, Brandt B, Berkun Y, et al. The relative contribution of environmental and genetic factors to phenotypic variation in familial Mediterranean fever (FMF)[J]. Gene, 2012, 491:260-263.
[7]Gibson KM, Hoffmann GF, Tanaka RD, et al. Mevalonate kinase map position 12q24[J]. Chromosome Res, 1997, 5:150.
[8]Houten SM, Frenkel J, Kuis W, et al. Molecular basis of classical mevalonic aciduria and the hyperimmunoglobulinaemiaD and periodic fever syndrome: high frequencyof 3 mutations in the mevalonate kinase gene[J]. J Inherit Metab Dis, 2000, 23:367-370.
[9]Mandey SH, Kuijk LM, Frenkel J, et al. A role for geranylgeranylation in interleukin-1beta secretion[J]. Arthritis Rheum, 2006, 54:3690-3695.
[10] Frenkel J, Houten SM, Waterham HR, et al. Clinical and molecular variability in childhood periodic fever with hyperimmunoglobulinaemia D[J]. Rheumatology (Oxford), 2001, 40:579-584.
[11] van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood[J]. Curr Rheumatol Rep, 2010, 12:101-107.
[12] Mandey SH, Schneiders MS, Koster J, et al. Mutational spectrum and genotype-phenotype correlations in mevalonate kinase deficiency[J]. Hum Mutat, 2006, 27:796-802.
[13] Simon A, Cuisset L, Vincent MF, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-igd and periodic fever syndrome: its application as a diagnostic tool[J]. Ann Intern Med, 2001, 135:338-343.
[14] Korppi M, Van Gijn ME, Antila K. Hyperimmuno-globulinemia Dand periodic fever syndrome in children. Review on therapy withbiological drugs and case report[J]. Acta Paediatr 2011; 100:21-25.
[15] Bodar EJ, Kuijk LM, Drenth JP, et al. On-demand anakinra treatment is effective in mevalonate kinasedeficiency[J]. Ann Rheum Dis, 2011, 70:2155-2158.
[16] Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinfammatory disorder[J]. Medicine (Baltimore), 2002, 81:349-368.
[17] Ravet N, Rouaghe S, Dodé C, et al. Clinical signifcance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene[J]. Ann Rheum Dis, 2006, 65:1158-1162.
[18] Borghini S, Tassi S, Chiesa S, et al. Clinical presentation and pathogenesis of cold-induced autoinfammatory disease in a family with recurrence of an NLRP12 mutation[J]. Arthritis Rheum, 2011, 63:830-839.
[19] Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Effcacy andsafety of anakinra therapy in pediatric and adult patients with the autoinfammatory Muckle-Wells syndrome[J]. Arthritis Rheum, 2011, 63:840-849.
[20] Hoffman HM, Throne ML, Amar NJ, et al. Effcacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies[J]. Arthritis Rheum, 2008, 58:2443-2652.
[21] Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome[J]. N Engl J Med, 2009, 360:2416-2425.
[22] van Duist MM, Albrecht M, Podswiadek M, et al. A new CARD15 mutation in Blau syndrome[J]. Eur J Hum Genet, 2005, 13:742-747.
[23] Lindor NM, Arsenault TM, Solomon H, et al. A new autosomal dominant disorder of pyogenic sterile arthritis, pyoderma gangrenosum, and acne: PAPA syndrome[J]. Mayo Clin Proc, 1997, 72:611-615.
[24] Dierselhuis MP, Frenkel J, Wulffraat NM, et al. Anakinra for flares of pyogenic arthritis in PAPA syndrome[J]. Rheumatology (Oxford), 2005, 44:406-408.
[25] Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome)[J]. J Med Genet, 2005, 42:551-557.
[26] Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist[J]. N Engl J Med, 2009, 360:2426-3247.
[27] Cochard M, Clet J, Le L, et al. PFAPA syndrome is not a sporadic disease[J]. Rheumatology (Oxford), 2010, 49:1984-1987.
[28] Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade[J]. Proc Natl Acad Sci U S A, 2011, 108:7148-7153.
[29] McGonagle D, Tan AL, Shankaranarayana S, et al. Management of treatment resistant inflammation of acute on chronic tophaceous gout with anakinra[J]. Ann Rheum Dis, 2007, 66:1683-1684.
[30] Gattorno M, Piccini A, Lasiglie D, et al. The pattern of response to anti-interleukin-1 treatment distinguishes two subsets of patients with systemic-onset juvenile idiopathic arthritis[J]. Arthritis Rheum, 2008, 58:1505-1515.
[31] Schroder K, Zhou R, Tschopp J. The NLRP3 inflammasome: a sensor for metabolic danger?[J]. Science, 2010, 327:296-300.
[32] Duewell P, Kono H, Rayner KJ, et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals[J]. Nature, 2010, 466:652.
[33] Zeft AS, Spalding SJ. Autoinflammatory syndromes: fever is not always a sign of infection[J]. Cleve Clin J Med, 2012, 79:569-581.
猜你喜欢
疯狂英语·新读写(2021年10期)2021-12-07
中老年保健(2021年10期)2021-08-24
天津医科大学学报(2021年3期)2021-07-21
中国生殖健康(2020年2期)2021-01-18
中华养生保健(2020年1期)2020-11-16
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
中国医药指南(2017年3期)2017-11-13
中成药(2017年3期)2017-05-17
医学研究杂志(2015年9期)2015-07-01
