
攀枝花苏铁自然保护区夏季PM2.5中二次有机物的研究
2014-06-23 07:47杨永琼代东决余志祥莫恒勤蒲金国邓仕槐
地球化学 2014年3期
赖 玮, 杨永琼, 李 黎*, 代东决, 余志祥, 莫恒勤, 蒲金国, 邓仕槐
攀枝花苏铁自然保护区夏季PM2.5中二次有机物的研究
赖 玮1, 杨永琼2, 李 黎1*, 代东决1, 余志祥2, 莫恒勤1, 蒲金国1, 邓仕槐1
(1. 四川农业大学 资源环境学院, 四川 成都 611130; 2. 四川攀枝花苏铁国家级自然保护区管理局, 四川 攀枝花 617000)
采用混合溶剂提取、N,O-双(三甲硅醚)-三氟乙酰胺(BSTFA)衍生化预处理和GC/MS分析技术, 对四川攀枝花苏铁国家自然保护区PM2.5中的大气二次有机气溶胶进行了定量检测, 探讨了研究区域内气溶胶中异戊二烯、α-/β-蒎烯光氧化产物、β-石竹酸及小分子羧酸和的浓度水平与变化规律, 并讨论了有机物占有机碳(OC)的比值。结果表明, 24 h PM2.5中, 异戊二烯光氧化产物、α-/β-蒎烯光氧化产物和β-石竹酸的平均浓度分别为51.2、16.1 和1.7 ng/m3; 苹果酸和2-羟基戊二酸的平均浓度分别为12.4 ng/m3和4.9 ng/m3。OC和元素碳(EC)的平均浓度分别为20.3 μg/m3和5.9 μg/m3。异戊二烯氧化产物、α-/β-蒎烯氧化产物及β-石竹烯氧化产物对OC的贡献率分别为1.63%、0.34%和0.36%。
PM2.5; 二次有机物; 异戊二烯光氧化产物; α-/β-蒎烯光氧化产物; 攀枝花苏铁自然保护区
0 引 言
森林植被向大气圈释放大量的挥发性有机化合物(volatile organic compounds, VOCs), 包括异戊二烯(isoprene)、单萜(monoterpene)、倍半萜(sesquiterpene)等[1]。从全球范围来看, 天然源VOCs的量远远超出人为源, 甚至在人类活动影响严重的地区, 人为源VOCs排放量也不及天然源VOCs[1–2]。
二次有机气溶胶(secondary organic aerosols, SOA)是指天然源VOCs和人为源VOCs与大气中主要的氧化剂, 如O3、NO3和OH自由基等, 反应生成难挥发性化合物, 经气-粒转化(gas-to-particle conversion)凝结至原生粒子(preexisting particles)上形成的亚微粒子[2]。 SOA能明显降低能见度, 造成阴霾天气; 并可作为云凝结核(cloud nucleation nuclei, CCN), 改变云的理化性质和寿命, 影响全球气候的变化。另外, 大气中SOA数量的增多还会对人体健康产生影响[2–3]。
目前, 关于天然源VOCs在大气中反应生成 SOA的研究报道主要集中在南美洲、欧洲和北美洲, 如巴西[4]、芬兰[5]、匈牙利[6]、德国[7]、美国[8–9]等。在我国, 涉及天然源SOA的研究则主要集中于东部几个森林地区[10-11]和亚热带城市广州[12]、香港[13], 西南林区仅有一例报道[14]。西南林区作为我国第二大林区, 是中国原始森林的主要分布地区之一, 它有着广阔的森林面积和多变的森林类型, 但是, 目前对该区域气溶胶中天然源SOA组分的研究却很少, 使得人们无法了解天然源SOA在西南森林地区的确切分布、浓度水平以及它与大气其他存在条件的相关作用。
四川攀枝花苏铁国家级自然保护区位于四川省攀枝花市西郊。攀枝花市地处川滇交界处, 属南亚热带为基带的立体气候。它气候干热, 夏季长, 年均气温20 ℃, 森林覆盖率58.97%, 是四川省年平均气温和总热量最高的地区。本研究选择于2011年7月在四川攀枝花苏铁自然保护区内采集PM2.5(大气中直径小于或等于2.5 μm的颗粒物)样本, 同时记录大气环境气象条件和监测大气污染气体(SO2、NO和O3)的浓度。拟通过实验室样品提取、硅醚衍生化与气相色谱/质谱联用分析方法检测气溶胶中SOA的浓度水平, 着重探讨异戊二烯氧化产物(isoprene oxidation products)、α-/β-蒎烯氧化产物(α-/β-pinene oxidation products)、β-石竹酸(β-caryophyllinic acid)在此区域的浓度范围和形成机制。
1 材料与方法
1.1 样品采集
1.1.1 颗粒物样品的采集
样品采集地点(26°36′N, 101°35′E)位于四川攀枝花苏铁国家自然保护区内, 保护区分布有目前世界上株数最多、面积最大、分布最集中的野生苏铁林。采样点海拔为1420 m。采样仪器为大气综合采样器(KC-6120, 青岛崂山电子有限公司), 流速为100 L/min。采样时间为2011年7月17~31日, 期间由于停电, 25日和26日中断了两天样品的采集。
采样滤膜为石英纤维滤膜(90 mm, 0.45 μm, Poll, USA)。采样前将滤膜置于550 ℃的马弗炉中焙烧 4 h。每天采集1个PM2.5的24 h样品(采集时间为20:00至次日19:30)和2个PM2.512 h样品, 日间和夜间样品的采集时间分别为当日的08:00至19:30和当日20:00至次日07:30。空白滤膜与上述采样方法相同, 只是采样时间为15 s。
1.1.2 气体样品的采集
SO2和NO的采集使用的是大气综合采样器(KC-6120, 青岛崂山电子有限公司)。吸收液分别采用0.04 mol/L四氯汞钾溶液和4 g/L对氨基苯磺酸+0.04 g/L-(1-萘基) 乙二胺盐酸混合溶液, 采样流量分别0.5 L/min和0.3 L/min。每种气体的采样持续时间为12 h, 采样时间段的划分和12 h颗粒物样本的采集相同。同时, 使用新西兰生产的Aeroqual 200高灵敏度臭氧测定仪测定环境中O3的浓度, 使用AM-19LQ Quantum meter(美国Avalon)测定光合有效辐射值(photosynthetically active radiation, PAR)。臭氧和光合有效辐射值均为日间1 h测定1次。采用Kestrel 4500手持式风速风向仪测量并记录环境温度、相对湿度、气压、风速和风向等气象数据。每日测定3次, 测定时间分别为当天的08:00、14:00和20:00。
1.2 样品的预处理
分别切取9~12 cm2石英膜于50 mL锥形瓶中, 加入800 ng D3-苹果酸(DMA, D3-malic acid, CDN isotopes, Canada)和600 ng甲基-β-D-木糖吡喃糖(MXP, methyl-β-D-xylanopyranoside, Sigma)作为内标; 然后, 加入20 mL二氯甲烷﹕甲醇=2﹕1的混合溶剂超声提取30 min, 重复3次。提取液放置澄清后, 取上清液转移至鸡心瓶中, 在旋转蒸发仪(RE 52-99, 上海)上旋转浓缩至1~2 mL。紧接着, 浓缩液经0.22 μm的PTFE针头过滤, 滤液转移至衍生化反应瓶中, 用柔和的高纯氮气吹干。最后, 加入40 μL BSTFA(-双(三甲硅醚)-三氟乙酰胺,bis (trimethylsilyl)-trifluroacetamide, BSTFA)+1%TMCS (三甲基氯硅烷, trimethylchlorosilane, TMCS)和无水吡啶(anhydrous pyridine)(2﹕1), 于恒温加热器中70 ℃反应1 h。
1.3 样品分析
1.3.1 GC/MS测定
气相色谱质谱仪(Agilent GC7890和MS5975)进样口温度为250 ℃, 不分流进样, 起始炉温50 ℃, 保留2 min, 以3 ℃/min的速度升温到200 ℃, 保留5 min, 30 ℃/min的速度升温到310 ℃, 保留2 min, 总运行时间为62.677 min。色谱柱型号为Rtx-5MS (30 m× i.d.0.25 mm, 膜厚0.25 μm)。采用选择离子扫描的方式对样品进行定量, 其中, MXP的定量碎片离子为204; 2-甲基甘油酸(2-methylglyceric acid)、2-甲基丁四醇(2-methyltetrols)的定量碎片离子为219; 异丁烯三醇(C5-alkene triols)的定量碎片离子为231; 苹果酸(malic acid)和D3-苹果酸的定量碎片离子分别为233和236。降蒎酸(norpinic acid)、3-甲基-1, 2, 3-丁三酸(3-methyl-1,2,3- butanetricarboxylic acid)和β-石竹酸的定量碎片离子分别为199,405和383; 3-羟基-4, 4-二甲基戊二酸(3-hydroxy-4,4-dimethylglutaric acid)的定量碎片离子为377; 3-羟基戊二酸(3-hydroxyglutaric acid)和2-羟基戊二酸(2-hydroxyglutaric acid)的定量碎片离子分别为259和247。
对于没有购买到完全一致标准品的化合物, 采用结构相近的标准品作为替代物(surrogate)进行定量, 如赤藻醇(erythritol)替代2-甲基甘油酸, 2-甲基丁四醇和异丁烯三醇; 苹果酸替代2-羟基戊二酸和3-羟基戊二酸; 蒎诺酸(pinonic acid)替代降蒎酸和β-石竹酸; 反-1, 4-环己二羧酸(trans-1,4-cyclohexane- dicarboxylic acid)替代3-甲基-1, 2, 3-丁三酸。环境样品浓度均减去空白样品浓度, 平行样之间的相对标准偏差小于10%。
1.3.2 OC-EC的测定
使用美国热/光碳分析仪(DRI Model 2001A)测定样品中的OC和EC浓度。具体的方法如下: 截取0.518 cm2的滤膜放在样品舟上, 并送入加热炉, 通过NIOSH (National Institute of Occupational Safety and Health)升温程序和激光透射法(thermo-optical transmission, TOT)进行测定, 总运行时间为13 min。
1.3.3 大气污染气体测定
采集SO2和NO后的吸收液分别按照中华人民共和国国家环境保护标准HJ 483-2009和HJ 479-2009进行显色测定。
1.4 实验试剂
D3-苹果酸(CDN Isotope, 加拿大, 99%);甲基-β- D-木聚吡喃糖(Sigma, 99%)。D(+)苹果酸(Fluka, 99%); 赤藻醇(山东, 99.8%); 反-1, 4-环己二羧酸(Aldrich, 99%); BSTFA+1%TMCS(Regis); 无水吡啶(Acros, 99%)。二氯甲烷(成都科龙化工试剂厂, A. R.); 甲醇(成都科龙化工试剂厂, A. R.); 二氯甲烷和甲醇均重蒸两次后使用。
2 结果与讨论
2.1 色谱图
在攀枝花苏铁国家自然保护区夏季大气PM2.5中, 我们定量检测到了异戊二烯光氧化产物、α-/β-蒎烯光氧化产物、β-石竹酸与和小分子羧酸(苹果酸、2-羟基戊二酸), 它们的GC/MS总离子流图见图1。
2.2 气溶胶中SOA的分析
表1显示的是采样期间PM2.5样品中的有机物平均浓度与浓度水平, 以及采样期间的大气污染气体浓度变化和环境气象条件。
2.2.1 异戊二烯光氧化产物
攀枝花苏铁自然保护区样品中一共检测出6种异戊二烯光氧化产物, 它们分别是: 2-甲基丁四醇(2-甲基苏阿醇(2-methylthreitol)和2-甲基赤藻醇(2-methylerythritol))、异丁烯三醇(顺式-2-甲基-1, 3, 4-三羟基-1-丁烯(cis-2-methyl-1,3,4-trihydroxy- 1-butene)、反式-2-甲基-1, 3, 4-三羟基-1-丁烯(trans-2-methyl-1,3,4-trihydroxy-1-butene)和3-甲基- 2, 3, 4-三羟基-1-丁烯(3-methyl-2,3,4-trihydroxy-1- butene))和2-甲基甘油酸。2-甲基苏阿醇和2-甲基赤藻醇之间存在着良好的线性关系(图2a)。2-甲基苏阿醇/2-甲基赤藻醇的平均浓度比值为0.3933, 换算成2-甲基赤藻醇占两种非对映异构体总和的比值(IF, isomeric fraction)为0.72。这一比值位于目前文献报道的关于气溶胶中2-甲基丁四醇IF值的范围(0.63~0.78)之间[15]。Kleindienst[16]和Nozière[17]分别从不同的烟雾箱实验中得出, 异戊二烯和OH·的气相光化学反应在无NO存在和有NO存在下, 以及酸性条件下异戊二烯的液相H2O2氧化, 生成的2-甲基丁四醇的IF值分别为0.61±0.02、0.76±0.01和0.90±0.02。以上的数据显示, 攀枝花苏铁自然保护区气溶胶中2-甲基丁四醇的形成机制和以往文献报道的研究类似, 2-甲基丁四醇的形成主要来自于OH·诱发的异戊二烯光化学反应, 即OH·进攻异戊二烯的双键, 形成异戊二烯羟基过氧自由基和异戊二烯羟基氢过氧化物(isoprene hydroxyl hydrogen peroxide, ISOPOOH), 后者进一步反应生成异戊二烯羟基环氧化物(isoprene expoxydiols, IEPOX), IEPOX极易在酸性颗粒物表面反应生成2-甲基丁四醇[18]。

图1 四川攀枝花苏铁国家级自然保护区大气气溶胶硅醚衍生化产物的总离子流图
1–2-甲基甘油酸; 2–异丁烯三醇(顺式-2-甲基-1, 3, 4-三羟基-1-丁烯、反式-2-甲基-1, 3, 4-三羟基-1-丁烯和3-甲基-2, 3, 4-三羟基-1-丁烯); 3–苹果酸/D3-苹果酸(IS); 4–2-甲基苏阿醇; 5–2-甲基赤藻醇; 6–2-羟基戊二酸/3-羟基戊二酸; 7–3-羟基-4, 4-二甲基戊二酸; 8–甲基-β-D-木聚吡喃糖(IS); 9–3-甲基-1, 2, 3-丁三酸

表1 采样期间攀枝花苏铁自然保护区大气PM2.5中OC、EC、有机物、痕量气体和气象条件的平均浓度与浓度范围
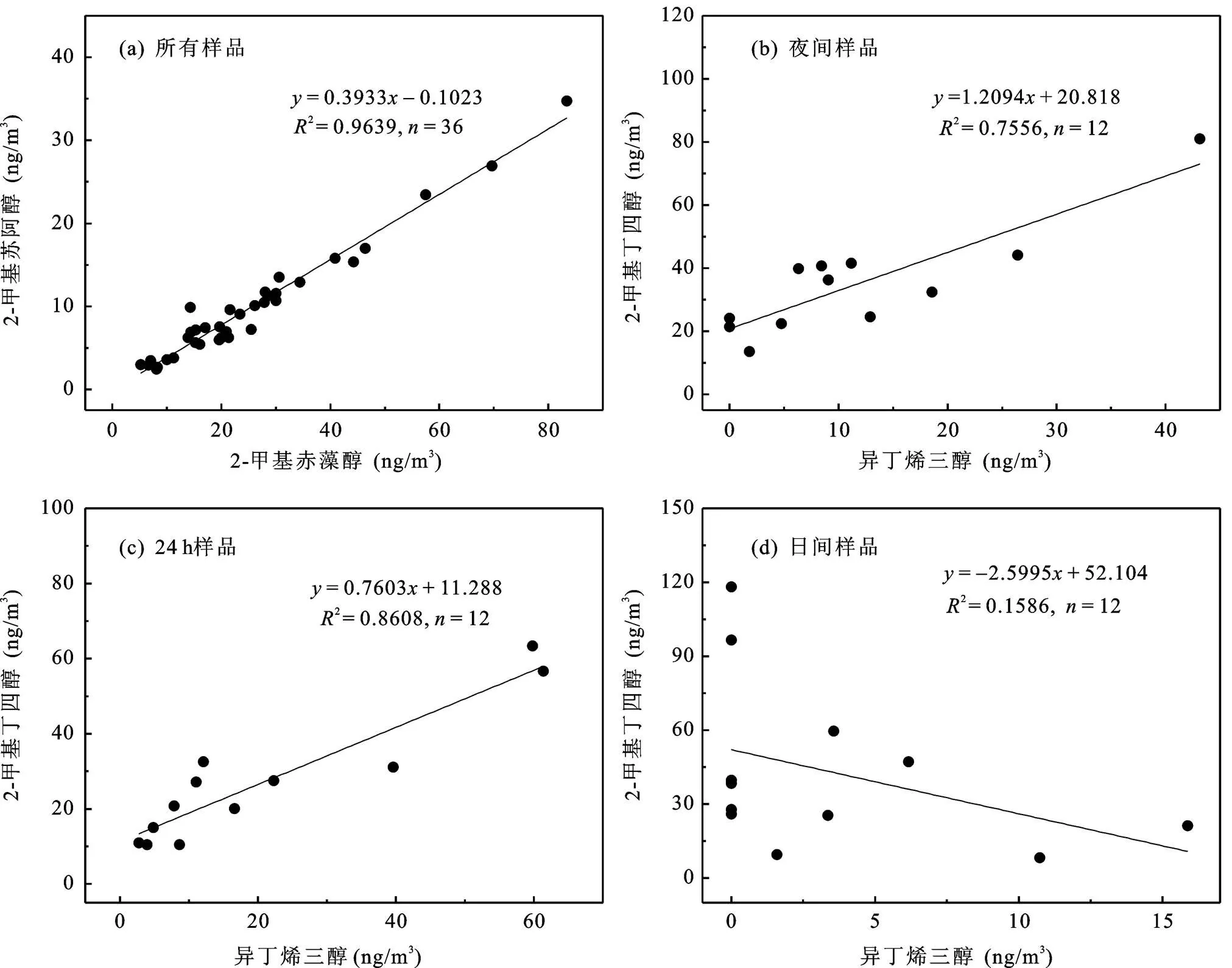
图2 2-甲基苏阿醇与2-甲基赤藻醇及2-甲基丁四醇与异丁烯三醇的相关性
此次检测出的2-甲基丁四醇的浓度明显高于一些地区的报道, 如德国西部混叶林[7](2003年夏季, 24 h PM2.5中2-甲基丁四醇平均浓度为15.2 ng/m3), 芬兰的Hyytiälä[5](2005年夏季, 2-甲基丁四醇在日间与夜间PM2.5中平均浓度分别为15.7ng/m3和10.3 ng/m3); 接近于匈牙利的K-puszta混叶林[6](2003年夏季, PM2.5中2-甲基丁四醇的日间和夜间平均浓度分别为44.1 ng/m3和20.2 ng/m3); 但低于巴西的Rondônia[4](2-甲基丁四醇在2002旱季的平均浓度为196 ng/m3), 中国泰山[12](2006年夏季, 2-甲基丁四醇日间和夜间平均浓度分别为98 ng/m3和123 ng/m3), 中国长白山[10](2007年夏季, PM2.5中2-甲基丁四醇日间和夜间平均浓度分别为130 ng/m3和113 ng/m3)。植物释放是异戊二烯最重要的来源, 植被覆盖率越高, 释放的异戊二烯越多; 温度越高, 异戊二烯释放量越大[1]; 前体物的增多将使得气溶胶中2-甲基丁四醇的生成量增多。此外, 颗粒物酸度的增强[18], 硫酸盐颗粒的增多[19], 均有利于提高2-甲基丁四醇的生成产率。
Wang[20]首次报道在亚马逊河热带雨林气溶胶样品中鉴定和发现了异丁烯三醇, 紧接着, 在芬兰[5]PM1和加利福尼亚[9]TSP气溶胶样品中均发现了异丁烯三醇, 此后, 在中国海南尖峰岭[10]、泰山[11]和雅安[14]等地也检测到了它们的存在。异丁烯三醇的形成过程被认为是IEPOX在酸催化下发生分子内重排而形成[20]。本研究发现, 2-甲基丁四醇与异丁烯三醇在夜间样品和24 h样品呈现较好的正相关关系(图2b、2c)。但是, 日间样品中2-甲基丁四醇与异丁烯三醇没有呈现相关性(图2d), 显示在2-甲基丁四醇和异丁烯三醇形成的过程中, 可能有因素的变化制约了两者形成的比例。烟雾箱[18]和环境样品的监测[14]则反映2-异丁烯三醇/甲基丁四醇的浓度比值受颗粒物的酸度影响明显, 强酸度下异丁烯三醇/2-甲基丁四醇的浓度比值明显降低。白天强烈的光化学反应生成了新生的硫酸根粒子, 在粒子表面强酸性条件下, 日间样品中异丁烯三醇/2-甲基丁四醇的浓度比值显著要比夜间和24 h样品中更低。
2-甲基甘油酸首先由Claeys课题组[21]在匈牙利平原的落叶/针叶森林PM2.5样品中发现。此后, Surratt[18]通过烟雾箱实验确证2-甲基甘油酸是异戊二烯光化学反应是高NO([NO]>500×10–9)条件下生成的主要SOA产物。其形成机制被解释为高NO条件下, 异戊二烯光化学反应首先生成2-甲基丙烯醛(methacrolein, MACR), 2-甲基丙烯醛进一步在OH·、O2与NO2的进攻下反应生成2-甲基丙烯酰基过氧硝酸酯(methacryloyl- peroxynitrate, MPAN), 而后降解生成2-甲基甘油酸。目前, 在不同地区气溶胶的监测中均发现细粒子中有2-甲基甘油酸[6,10,11,14]的存在。
2.2.2 α-/β-蒎烯光氧化产物
α-/β-蒎烯是自然界中排放量最大的单萜类化合物。本研究检测出了四种α-/β-蒎烯的光氧化产物, 即降蒎酸、3-羟基戊二酸、3-羟基-4, 4-二甲基戊二酸和3-甲基-1, 2, 3-丁三酸, 它们在24 h、日间和夜间样品中的总平均浓度分别为16.1、22.9和22.2 ng/m3。此浓度水平高于珠江三角洲郊区的6.61 ng/m3[11]; 接近于德国西部混交林PM2.5中α-/β-蒎烯氧化产物的平均浓度25.6 ng/m3[11]、中国雅安白马泉的监测数据[22](24 h, 日间, 夜间平均浓度分别为14.4、10.6和14.8 ng/m3)和中国泰山山顶[10](日间和夜间平均浓度分别为33 ng/m3和27 ng/m3); 但明显低于芬兰的Hyytiälä[5]的α-/β-蒎烯氧化产物监测结果(24 h平均浓度65ng/m3)。
研究显示, 降蒎酸是α-/β-蒎烯光氧化反应的主要初级颗粒相氧化产物, 在O3与OH· 存在的烟雾箱试验中可以产生[5,7]。此外,3-羟基戊二酸、3-羟基- 4, 4-二甲基戊二酸和3-甲基-1,2,3-丁三酸均为更老化的α-/β-蒎烯光氧化产物。这其中, Claeys[23]首先确认了3-羟基戊二酸和3-羟基-4,4-二甲基戊二酸, Szmigielski[24]确认了3-甲基-1,2,3-丁三酸, 他们发现这三种化合物均能在NO存在下通过紫外辐射α-蒎烯产生。Ding.[12]研究发现, 广州PM2.5中3-羟基戊二酸、3-羟基-4, 4-二甲基戊二酸和3-甲基-1, 2, 3-丁三酸三者之间相关性良好。本研究也发现24 h PM2.5中3-羟基戊二酸、3-羟基-4, 4-二甲基戊二酸和3-甲基-1, 2, 3-丁三酸彼此之间的相关性良好(图3a、3b、3c)。Zhang[25]报道环境中OH·含量是决定气溶胶中3-甲基-1,2,3-丁三酸水平的重要因子, 以上相关性的吻合说明OH·含量可能是决定气溶胶中老化的α-/β-蒎烯光氧化产物形成的关键因素。
2.2.3 倍半萜光氧化产物
β-石竹酸是倍半萜的光氧化产物, 本研究中, β-石竹酸在24 h、日间和夜间PM2.5样品中的平均浓度分别为1.7、3.0和2.2 g/m3, 高于中国珠江三角洲地区(平均浓度为0.54 ng/m3)[12]; 但明显低于中国泰山(日间和夜间平均浓度均为12 ng/m3)[11]。研究中还发现夜间样品中β-石竹酸和降蒎酸之间有很好的相关性(图3d)。Jaoui[26]曾报道β-石竹酸与降蒎酸在黑暗环境中的形成机制相似, 主要是通过O3与VOCs氧化生成。
2.2.4 小分子羧酸
苹果酸是非饱和脂肪酸的次级产物, 是气溶胶中天然源和人为源二次有机组分的多步降解产物; 苹果酸是自然环境中常见的小分子羧酸, 在细粒子中最丰富。本研究中苹果酸在24 h、日间和夜间样品中的平均浓度分别为12.4、8.5和21.3 ng/m3。与苹果酸类似, 2-羟基戊二酸也被认为是非饱和脂肪酸的光降解产物[27], 其在采样期间24 h、日间和夜间样品中的平均浓度分别为4.9、4.7和7.8 ng/m3。本研究发现2-羟基戊二酸和3-羟基戊二酸之间的相关性(2=0.3485,=36, 图4)明显不如在雅安白马泉监测的结果[14], 显示2-羟基戊二酸和3-羟基戊二酸之间在此次采样期间的确存在着来源上的不同。
2.3 SOA变化规律与SOA占OC的比例
烟雾箱实验显示, 在反应过程中通入SO2可显著提高异戊二烯、单萜和倍半萜光氧化生成SOA的产量[28–29]。同时, NO对SOA的生成也有明显的影响。Kroll[30]研究发现, 在高浓度NO下, 增加NO浓度会导致SOA的急剧下降, 然而, 在低NO下, 增加NO会引发SOA生成量的增加。采样期间痕量气体及气象条件的平均浓度及浓度水平如表1所示。24 h SO2和NO的平均浓度分别为65.1和8.6μg/m3, 日间臭氧的平均浓度50.1 μg/m3。采样期间SO2的浓度变化范围为17.3~169.9 μg/m3, SO2的浓度呈现昼高夜低。采样点附近是攀钢集团有限公司, 另外与很多煤矿相邻, 这是导致SO2浓度过高的原因。较高的SO2环境浓度可提高植物排放VOC生成SOA的产率。此外, 采样期间温度、湿度、风速及光合有效辐射值的平均值分别为33.4 ℃、34%、2.2 m/s和1231 μmol/(m2·s), 持续的高温和充足的光照也可促进气溶胶中SOA组分的生成。

图3 24 h样品α-/β-蒎烯氧化产物及夜间样品β-石竹酸和降蒎酸的相关性

图4 2-羟基戊二酸和3-羟基戊二酸之间的相关性
影响SOA昼夜变化的因素有光化学反应、大气边界层的运动以及气粒间的两相分配。日间光照强烈, 有利于植物排放异戊二烯、单萜、倍半萜等SOA的前体物, 因此, 天然源SOA, 特别是生成速度较快, 反应路径短的一些SOA产物通常表现为日间浓度高于夜间浓度, 如2-甲基丁四醇、降蒎酸、β-石竹酸(表1)。而大气边界层的运动则和光化学反应的影响相反, 夜间大气污染物向地表的汇集会导致夜间一些SOA成分浓度的升高。此外, 夜间温度的降低更有利于SOA成分向颗粒相凝聚, 相比于日间, 夜间颗粒相的酸度降低, 尤其利于一些有机酸类的SOA组分向颗粒相分配[12]。SOA在采样期间的时间变化趋势见图5。此次野外研究, 除了2-甲基丁四醇、降蒎酸、β-石竹酸之外, 其他的SOA标志物均表现为平均夜间浓度高于平均日间浓度(表1)。

图5 采样期间气象条件、污染气体及SOA的时间变化趋势
D代表日间样品,N代表夜间样品, PAR代表光合有效辐射值(photosynthetically active radiation)
本研究中, OC和EC的24 h的平均浓度分别为22.7 μg/m3和6.4 μg/m3(表1), 明显高于匈牙利的K-puszta[6](24 h OC和EC平均浓度分别为4.2 μg/m3和0.2 μg/m3), 芬兰的Hyytiälä[5](24 h OC和EC中值浓度分别为2.2 μg/m3和0.04 μg/m3); 接近于中国泰山[11](日间和夜间OC平均浓度分别为19 μg/m3和21 μg/m3)和中国珠江三角洲[12](OC、EC平均浓度分别为19.3 μg/m3和3.55 μg/m3)的浓度。24 h PM2.5中OC/EC比值为3.55, 接近于上海郊区佘山[31](夏季OC/EC比值为2.4)、广州郊区荔湾[32](夏季OC/EC比值为3.5)、香港郊区Hok Tsui[32](夏季OC/EC比值为3.8)和雅安四川农业大学校区[14](夏季OC/EC比值为5.0)。攀枝花市为一重要工业城市, 采样期间, 有货车在采样点山下运送煤炭, 昼夜不停, 煤灰粉尘和汽车的尾气排放会导致EC浓度的增加; 同时由于汽车尾气的排放, 夜间NO的浓度甚至高于日间浓度。
采样期间大气PM2.5中SOA占OC的比值如表2所示。异戊二烯、α-/β-蒎烯及倍半萜氧化产物对OC贡献率的计算采用Kleindienst[8]提出的方法(异戊二烯、α-/β-蒎烯氧化产物和β-石竹酸占OC的质量分数分别为0.155、0.231和0.023)。从表2中可以看出, 三类SOA对OC的贡献之和为2.33%, 其中夜间的总贡献要高于日间, 可能是夜间人为污染排放量有所降低, 使得OC总量有明显下降所致。
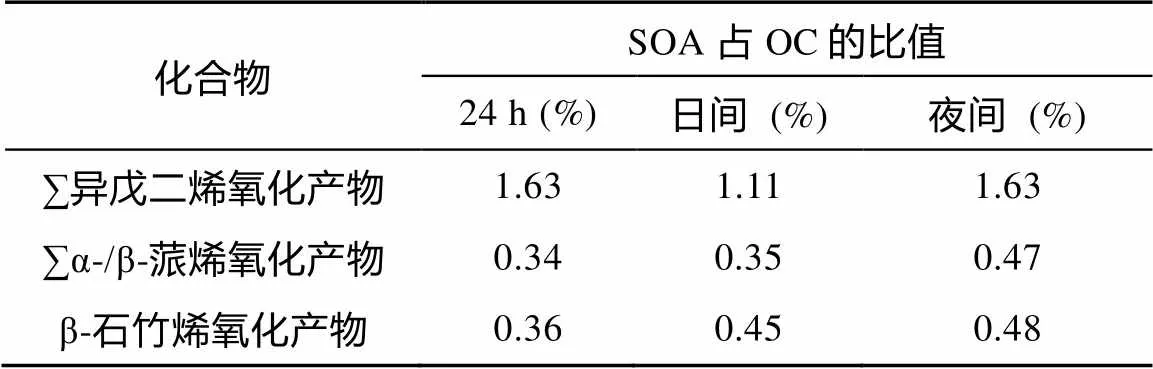
表2 攀枝花苏铁自然保护区大气PM2.5中天然源SOA占OC的比值
3 结 论
研究选择于2011年夏季在攀枝花苏铁国家自然保护区采集颗粒物样本, 测定其大气中二次有机气溶胶组分的含量。24 h PM2.5样品中, 检测到的总异戊二烯光氧化产物和总α-/β-蒎烯光氧化产物的浓度分别为51.2 ng/m3和16.1 ng/m3, 异戊二烯、α-/β-蒎烯和石竹烯氧化产物对OC贡献率分别为1.63%、0.34%和0.36%。夜间和24 h样品中2-甲基丁四醇与异丁烯三醇之间相关性良好, 但是日间样品中没有表现出相关性, 推测是由于受到环境中SO2浓度的影响。同时, 24 h样品中α-/β-蒎烯光氧化产物 3-羟基戊二酸、3-羟基-4, 4-二甲基戊二酸和3-甲基- 1, 2, 3-丁三酸三者之间有较好的相关性, 夜间样品中β-石竹酸和降蒎酸相关性良好, 以上相关性揭示SOA在形成过程中可能受到了环境中OH·和O3水平的制约。
感谢上海大学环境污染与健康研究所冯加良老师在OCEC测定中给予的帮助; 同时感谢四川省雅安市环境监测站的刘子芳站长和王松涛工程师在GC/MS测定上给予的支持。
[1] Guenther A, Hewitt C N, Erickson D, Fall R, Geron C, Graedel T, Harley P, Klinger L, Lerdau M, Mckay W A, Pierce T, Scholes B, Steinbrecher R, Tallamraju R, Taylor J, Zimmerman P. A global model of natural volatile organic compound emissions[J]. J Geophys Res, 1995, 100(D5): 8873–8892.
[2] 汪午, 王省良, 李黎, 张东平, 王扬君, 盛国英, 傅家谟. 天然源二次有机气溶胶的研究进展[J]. 地球化学, 2008, 37(1): 77–86.
Wang Wu, Wang Sheng-liang, Li Li, Zhang Dong-ping, Wang Yang-jun, Sheng Guo-ying, Fu Jia-mo. Advances in biogenic secondary organic aerosols[J]. Geochimica, 2008, 37(1): 77–86 (in Chinese with English abstract).
[3] Pathak R K, Yao X H, Lau A K H, Chang C K. Acidity and concentrations of ionic species of PM2.5in Hong Kong[J]. Atmos Environ, 2003, 37(8): 1113–1124.
[4] Claeys M, Kourtchev I, Pashynska V, Vas G, Vermeylen R, Wang W, Cafmeyer J, Chi X, Artaxo P, Andreae M O, Maenhaut W. Polar organic marker compounds in atmospheric aerosols during the LBA-SMOCC 2002 biomass burning experiment in Rondônia, Brazil: Sources and source processes, time series, diel variations and size distributions[J]. Atmos Chem Phys, 2010, 10(19): 9319–9331.
[5] Kourtchev I, Ruuskanen T M, Keronen P, Sogacheva L, Dal Maso M, Reissell A, Chi X, Vermeylen R, Kulmala M, Maenhaut W, Claeys M.Determination of isoprene and α-/β-pinene oxidation products in boreal forest aerosols from Hyytiälä, Finland: Diel variations and possible link with particle formation events[J]. Plant Biol, 2008, 10(1): 138–149.
[6] Ion A C, Vermeylen R, Kourtchev I, Cafmeyer J, Chi X, Gelencsér A, Maenhaut W, Claeys M. Polar organic compounds in rural PM2.5aerosols from K-puszta, Hungary, during a 2003 summer field campaign: Sources and diel variations[J]. Atmos Chem Phys, 2005, 5(7): 1805–1814.
[7] Kourtchev I, Warnke J, Maenhaut W, Hoffmann T, Claeys M. Polar organic marker compounds in PM2.5aerosol from a mixed forest site in western Germany[J]. Chemosphere, 2008, 73(8): 1308–1314.
[8] Kleindienst T E, Jaoui M, Lewandowski M, Offenberg J H, Lewis C W, Bhave P V, Edney E O. Estimates of the contributions of biogenic and anthropogenic hydrocarbons to secondary organic aerosol at a southeastern US location[J]. Atmos Environ, 2007, 41(37): 8288–8300.
[9] Cahill T M, Seaman V Y, Charles M J, Holzinger R, Goldstein A H. Secondary organic aerosols formed from oxidation of biogenic volatile organic compounds in the Sierra Nevada Mountains of California[J]. J Geophys Res, 2006, 111: D16312, doi: 101029/2006 JD007178.
[10] Wang W, Wu M H, Li L, Zhang T, Liu X D, Feng J L, Li H J, Wang Y J, Sheng G Y, Claeys M, Fu J M. Polar organic tracers in PM2.5aerosols from forests in eastern China[J]. Atmos Chem Phys, 2008, 8(3): 7507–7518.
[11] Fu P, Kawamura K, Kanaya Y, Wang Z F. Contributions of biogenic volatile organic compounds to the formation of secondary organic aerosols over Mt.Tai, Central East China[J].Atmos Environ, 2010, 44(38): 4817–4826.
[12] Ding X, Wang X M, Zheng M. The influence of temperature and aerosol acidity on biogenic secondary organic aerosol tracers: Observations at a rural site in the central Pearl River Delta region, South China[J]. Atmos Environ, 2011, 45(6): 1303–1311.
[13] Hu D, Bian Q, Li T W Y, Lau A K H, Yu J Z. Contributions of isoprene, monoterpenes, β-caryophyllene, and toluene to secondary organic aerosols in Hong Kong during the summer of 2006[J]. J Geophys Res, 2008, 113, D22206, http://dx.doi. org/10.1029/2008JD010437.
[14] Li L, Dai D J, Deng S H, Feng J L, Zhao M, Wu J, Liu L, Yang X H, Wu S S, Qi H, Yang G, Zhang X H, Wang Y J, Zhang Y Z. Concentration, distribution and variation of polar organic aerosol tracers in Ya’an, a middle-sized city in western China[J]. Atmos Res, 2013, 120: 29–42.
[15] Zhang Z S, Engling, G, Chan, C Y, Yang Y H, Lin M, Shi S, He J, Li Y D, Wang X M. Determination of isoprene-derived secondary organic aerosol tracers (2-methyltetrols) by HPAEC-PAD: Results from size-resolved aerosols in a tropical rainforest[J]. Atmos Environ, 2013, 70: 468–476.
[16] Kleindienst T E, Lewandowski M, Offenberg J H, Jaoui M, Edney E O. The formation of secondary organic aerosol from the isoprene + OH reaction in the absence of NO[J]. Atmos Chem Phys, 2009, 9(2): 6541–6558.
[17] Nozière B, González Nélida J D, Borg-Karlson A K, Pei Y X, Redeby J P, Krejci R, Dommen J, Prevot A S H, Anthonsen T. Atmospheric chemistry in stereo: A new look at secondary organic aerosols from isoprene[J]. Geophys Res Lett, 2011, 38: L11807, doi: 10.1029/ 2011GL047323.
[18] Surratt J D, Chan A W H, Eddingsaas N C, Chan M N, Loza C L, Kwanb A J, Hersey S P, Flagan R C, Wennberg P O, Seinfeld J H. Reactive intermediates revealed in secondary organic aerosol formation from isoprene[J]. Proc Natl Acad Sci USA, 2010, 107(15): 6640–6645.
[19] Liang L L, Engling G, Duan F K, Cheng Y, He K B. Characteristics of 2-methyltetrols in ambient aerosol in Beijing, China[J]. Atmos Environ, 2012, 59: 376–381.
[20] Wang W, Kourtchev I, Graham B, Cafmeyer J, Maenhaut W, Claeys M. Characterization of oxygenated derivatives of isoprene related to 2-methytetrols in Amazonian aerosols using trimethylsilylation and gas chromatography/ion trap mass spectrometry[J]. Rapid Commun Mass Spectrom, 2005, 19(10): 1343–1351.
[21] Claeys M, Wang W, Ion A C, Kourtchev I, Gelencser A, Maenhaut W. Formation of secondary organic aerosols from isoprene and its gas-phase oxidation products through reaction with hydrogen peroxide[J]. Atmos Environ, 2004, 38(25): 4093–4098.
[22] 代东决, 李黎, 刘子芳, 赵敏, 冯加良, 赖玮, 曾燕梅, 周宇, 刘露云, 邓仕槐. 白马泉风景区夏季大气PM2.5中二次有机物的初步研究[J]. 环境科学, 2012, 33(4): 1063–1072.
Dai Dong-jue, Li Li, Liu Zi-fang, Zhao Min, Feng Jia-liang, Lai Wei, Zeng Yan-mei, Zhou Yu, Liu Lu-yun, Deng Shi-huai. Secondary organic tracers in summer PM2.5aerosols from Baima Spring Scenic Area, Ya’an, Sichuan Province[J]. Environ Sci, 2012, 33(4): 1063–1072 (in Chinese with English abstract).
[23] Claeys M, Szmigielski R.Hydroxydicarboxylic acids: novel markers for secondary organic aerosol from the photooxidation of α-pinene[J]. Environ Sci Technol, 2007, 41(5): 1628–1634.
[24] Szmigielski R, Surrat J D, Gómez-González Y, Kourtchev I, Vermeylen R, Blockhuys F, Jaoui M, Kleindienst T E, Lewandowski M, Offenberg J H, Edney E O, Seinfeld J H, Maenhaut W, Claeys M. 3-methyl-1,2,3-butanetricarboxylic acid: An atmospheric tracer for terpene secondary organic aerosol[J]. Geophys Res Lett, 2007, 34(24): L24811, doi: 10.1029/2007GL031338.
[25] Zhang Y Y, Müller L, Winterhalter R, Moortgat G K, Hoffmann T, Pöschl U. Seasonalcycle and temperature dependence of pinene oxidation products, dicarboxylic acids and nitrophenols in fine and coarse air particulate matter[J]. Atmos Chem Phys, 2010, 10(16): 7859–7873.
[26] Jaoui M, Lewandowski M, Kleindienst T E, Offenberg J H, Edney E O. β-caryophyllinic acid: An atmospheric tracer for β-caryophyllene secondary organic aerosol[J]. J Geophys Res Lett, 2007, 34(5): 1–4.
[27] Gómez-González Y, Surratt J D, Cuyckens F, Szmigielski R. Characterization of organosulfates from the photooxidation of isoprene and unsaturated fatty acids in ambient aerosol using liquid chromatography/(-) electrospray ionization mass spectrometry[J]. J Mass Spectrom, 2008, 43(3): 371–382.
[28] Edney E O, Kleindienst T E, Jaoui M, Lewandowski M, Offenberg J H, Wang W, Claeys M. Formation of 2-methyltetrols and 2-methylglyceric acid in secondary organic aerosol from laboratory irradiated isoprene/NO/ SO2/air mixtures and their detection in ambient PM2.5samples collected in the eastern United States[J]. Atmos Environ, 2005, 39(29): 5281–5289.
[29] Kleindienst T E, Edney E O, Lewandowski M, Offenberg J H, Jaoui M. Secondary organic carbon and aerosol yields from the irradiations of isoprene and α-pinene in the presence of NOand SO2[J]. Environ Sci Technol, 2006, 40(12): 3807–3812.
[30] Kroll J H, Murphy S M, Flagan R C, Seinfeld J H.Secondary organic aerosol formation from isoprene photooxidation[J]. Environ Sci Technol, 2006, 40(6): 1869–1877.
[31] Feng J L, Chan C K, Fang M, Hu M, He L, Tang X. Characteristics of organic matter in PM2.5in Shanghai[J]. Chemosphere, 2006, 64(8): 1393–1400.
[32] Duan J C, Tan J H, Cheng D X, Bi X H, Deng W J, Sheng G Y, Fu J M, Wong M H. Sources and characteristics of carbonaceous aerosol in two largest cities in Pearl River Delta Region, China[J]. Atmos Environ, 2007, 41(14): 2895–2903.
Secondary organic tracers in summer PM2.5aerosols from Sichuan Panzhihua Cycad NationalNature Reserve
LAI Wei1, YANG Yong-qiong2, LI Li1*, DAI Dong-jue1, YU Zhi-xiang2, MO Heng-qin1, PU Jin-guo1and DENG Shi-huai1
1. College of Resources and Environmental Sciences, Sichuan Agricultural University, Chengdu 611130, China; 2. Sichuan Panzhihua Cycad National Nature Reserve, Panzhihua 617000, China
PM2.5aerosol samples, collected during the summer of 2011 in Sichuan Panzhihua Cycad National Nature Reserve, were extracted with mixed solution, derivatized with N,O-bis-(trimethylsilyl) trifluoroacetamide (BSTFA) and analyzed by gas chromatography/mass spectrometry (GC/MS). The objectives of this study were to examine the time series of photooxidation products of isoprene, α-/β-pinene and β-caryophyllene and of some small molecular carboxylic acids, to determine their concentrations and estimate their contributions to aerosol organic carbon (OC). The results showed that average concentrations of total isoprene and α-/β-pinene oxidation products and β-caryophyllinic acid in 24 h PM2.5aerosol samples were 51.2, 16.1 and 1.7 ng/m3, respectively. Average levels of malic acid and 2-hydroxyglutaric acid were 12.4 ng/m3and 4.9 ng/m3, and the mean concentrations of OC and elemental carbon (EC) were 20.3 μg/m3and 5.9 μg/m3, respectively. Contributions of isoprene, α-/β-pinene and β-caryophyllene oxidation products to OC were 1.63%, 0.34% and 0.36% during sampling campaign, respectively.
PM2.5; secondary organic tracers; isoprene oxidation products; α-/β-pinene oxidation products; Sichuan Panzhihua Cycad NationalNature Reserve
P597
A
0379-1726(2014)03-0197-11
2013-04-15;
2013-06-25;
2013-07-30
国家自然科学基金(41073101); 四川省教育厅青年基金项目(11ZB065)
赖玮(1989–), 女, 硕士研究生, 主要研究方向为大气环境学。E-mail: llww1989@163.com
LI Li, E-mail: lili@sicau.edu.cn; Tel: +86-28-86291132
猜你喜欢
辐射防护通讯(2019年3期)2019-04-26
四川环境(2019年6期)2019-03-04
中成药(2018年2期)2018-05-09
成都信息工程大学学报(2017年2期)2017-11-09
新乡学院学报(2016年6期)2016-12-01
高原山地气象研究(2016年2期)2016-11-10
当代化工研究(2016年9期)2016-03-20
微生物学杂志(2016年6期)2016-03-07
橡塑技术与装备(2016年21期)2016-02-25
