
AuX(X=O,S)低电子态的理论研究
2014-10-18 05:28梁艳妮
物理化学学报 2014年8期
梁艳妮 王 繁
(1四川大学化学学院,成都 610065;2四川大学原子与分子物理研究所,成都 610065)
1 引言
金的价电子组态为5d106s1,与强碱性原子具有相似的电子结构.但是,非常强的相对论效应使金在贵金属中表现出不寻常的物理和化学性质.相对论效应使Au原子的6s轨道能量降低,导致反常的电离能(IP=9.225 eV)和电子亲和能(EA=2.309 eV).相对论效应同时会使5d轨道半径增大,能量升高,致使s-d能差减小而引起其物理和化学性质的变化.1-3因此,金作为金属却同样表现出强的共价键特性以及独特的化学性质.近年来,金在催化及纳米技术中的广泛应用,尤其是在低温催化CO氧化反应中的卓越表现,吸引众多课题组开展金氧化物团簇的研究工作,而最简单的团簇分子AuO和AuO-的性质对阐明和理解这些团簇的结构和性质非常重要,有关此类一氧化物已有大量的实验以及理论计算研究.另外,Au―S键在自组装单分子膜形成中具有重要作用,4但是目前有关AuS和AuS-的光谱研究较少.早期的电子发射光谱报道了AuO分子的振动频率为699 cm-1.51999年的AuO电子光谱实验对应于16O/18O的振动频率分别为619.2和586.9 cm-1.6直到2004年,Gantefor研究组7完成了AuO-在193 nm的光电子能谱实验.几乎在同时,Lineberger等8报道了AuO-和AuS-在364 nm的高分辨率光电子能谱实验,该实验得到AuO和AuS的EA分别是(2.374±0.007)eV和(2.469±0.006)eV,并清晰地看到了2Π态的旋轨耦合分裂,AuO和AuS的2Π1/2态和2Π3/2态之间的旋轨分裂能分别为(0.179±0.010)eV和(0.159±0.007)eV.另外,该实验光谱Franck-Condon分析表明,AuO-的基态1Σ+态的键长比AuO的2Πi态的平均键长短,然而AuS-的1Σ+态的键长却比AuS的2Πi态的平均键长更长.此键长关系反映了这两个体系阴离子和中性分子之间不同程度的相对论效应.8之后也有研究组报道了AuO激发态4Σ-的近红外电子光谱9,10和基态2Π3/2的转动光谱.11以上实验只是围绕AuX基态和第一激发态所做的光谱分析.直到近期,Wang研究组12报道了AuO-和AuS-的光电子能谱实验,并通过对AuO和AuS低电子态的理论计算指认得到光电子能谱.
在理论研究AuO和AuS低电子态方面,由于涉及Au原子,必须考虑相对论效应才能得到合理的结果.相对论效应分为标量相对论效应和旋轨耦合(SOC)效应.不考虑SOC时常用的相对论有效势与非相对论有效势在数学形式上完全一致,如果要考虑SOC效应则计算量会明显增加.Legge,13Wu14和Su15等采用密度泛函方法(DFT),在不考虑SOC效应下计算了AuXq(q=1,0,-1)基态分子的键长、频率、EA、IP和解离能(DE).其计算结果依赖于所使用的泛函,采用BP8616,17和BLYP16,18泛函得到的数据更接近实验和理论计算结果.Lineberger等8则采用了包括完全单重、双重激发和不完全三重激发的CCSD(T)方法计算了AuO-和AuS-的键长和IP,用多组态相互作用(MRCI)方法在态平均的完全活性空间自洽场(CASSCF)基础上计算包含SOC效应的AuO和AuS基态2Π的旋轨耦合分裂态2Π3/2和2Π1/2的光谱常数.之后,O′Brien等10则采用全活性空间二阶微扰方法(CASPT2),基于CASSCF(12,15)计算了AuO的势能曲线,此计算在考虑了相关能的电子态之间计算旋轨耦合作用,19计算了4Σ-、2Σ-、4Π和2Π态的旋轨耦合分裂态的光谱常数和垂直激发能.他们得到的2Π3/2态键长为0.1861 nm,与实验值0.1849 nm11吻合较好,第一激发态2Π1/2所对应的绝热激发能为0.173 eV,也与先前的实验值(0.179±0.010)eV吻合.8他们的结果显示2是第二个激发态,绝热激发能为1.406 eV,后面依次为态和.迄今为止有关AuO和AuS分子基态和低激发态光谱常数计算较为全面的是Wang等12基于态平均CASSCF(17,10)计算电子态之间的SOC作用.此计算与他们得到的实验光谱解析吻合非常好.理论研究AuX(X=O,S)的低电子态一般需要用多参考态方法描述,这主要由于需要计算开壳层分子的几个低电子态之间的SOC作用,要求这几个低电子态的波函数具有正确的空间和自旋对称性,而这很难用传统的计算激发能的单参考态方法处理.值得注意的是Liu小组20最近发展的含SOC的含时密度泛函(TD-DFT)二分量的相对论方法(sf-X2C-S-TD-DFT-SOC)应该可用来处理这个问题.但是,多参考态方法是非“黑箱”方法,对使用者有较高的要求,需要对所研究体系有比较深入的认识,才能选择合适的参考态从而得到合理的结果,而且计算结果依赖于所选参考态.此外,增加活性空间也会使计算量大大增加.
事实上,对于一些多组态特性显著的电子态,如果能够从一个可用单行列式波函数合理描述的电子态出发,通过自旋反转跃迁、电离或者添加一个乃至多个电子得到所研究的状态,这样就能够避免参考态的选取.Krylov21首先提出采用自旋反转跃迁的方法研究多参考态体系,并在CI以及运动方程耦合簇(EOM-CC)计算电子激发的方法中实现.EOM-CC计算单电离或电子亲和方法则在发展初期就已经被用于研究多参考态体系.22,23我们课题组22,24近期发展了一套含SOC的EOM-CC计算电离能的方法(EOMIP-SOC-CC).在EOMIP-CC方法中,首先用CC方法计算比目标态多一个电子的闭壳层参考态的能量和波函数,再通过求解EOMIP方程计算目标态的能量和波函数.以前的计算结果25,26证实了在CC计算中就考虑SOC(SOC-CC)的方法能精确地描述重元素分子的SOC效应,这是由于CC方法中单激发算符能够很好地描述轨道驰豫.此外,EOMIP方法选用闭壳层体系作为参考态的做法使体系波函数能完全避免自旋污染,而这对于描述SOC尤其重要.因为使用非自旋纯态的波函数计算SOC效应会导致原本不相互作用的态之间由于SOC而相互耦合,造成错误的能级分裂.另一方面,我们的方法只在求解CC方程和EOMIP方程时考虑SOC,因此在相关能计算中使用的是实自旋轨道,这比在SCF计算时就考虑SOC的方法效率更高.目前常用的EOMIP-CC方法都是基于CCSD级别,主要适用从参考态电离一个电子而不激发另一个电子所得到的电子态,对于这种电子态所得到的电离能误差一般在0.1-0.3 eV.此外,如果CCSD方法能够合理地描述参考态,而目标态可以通过从参考态拿走一个电子而不激发另一个电子获得,EOMIP-CCSD方法也能适用于描述多参考态体系,我们以前的工作27,28证实了这种计算的可靠性.在CC方法中,要得到高精度的计算结果往往需要考虑三重激发的贡献,采用微扰法考虑三重激发贡献的EOMIP-CC方法能提高EOMIP-CCSD方法的计算精度29,30而且计算量适中,但是这些方法尚未在含旋轨耦合的EOMIP-CC方法上实现.
本文中我们采用含SOC的EOMIP-CCSD方法计算开壳层体系AuO和AuS低电子态的光谱常数以及AuO-和AuS-到这些态所对应的垂直电离能(VIP)和绝热电离能(AIP),并与以前的理论和实验结果比较,以考察我们的方法在研究此类原本需要用多参考态方法处理的含SOC体系的精确度.
2 基本理论和计算细节
在耦合簇理论中,CCSD方法31的总能量为

其中T=T1+T2为激发算符,其定义如下
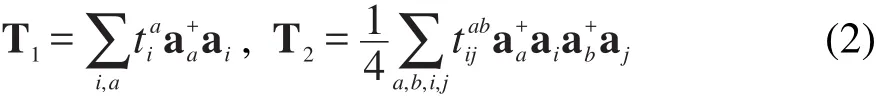
上面式子中Φ0为HF行列式波函数,H为体系的哈密顿量,i,j,…和a,b,…分别对应波函数Φ0的占据轨道和空轨道,算符和ai分别表示将其作用于Φ0时电子的产生和湮灭算符,和分别表示激发算符T1和T2的振幅,其中T1和T2满足以下方程:

为了在考虑SOC时计算开壳层分子,我们在闭壳层体系含SOC的CC基础上实现了运动方程耦合簇方法(EOM-CC)34计算电离能的办法,即EOMIPSOC-CCSD方法,24该方法适用于计算比闭壳层体系少一个电子的开壳层体系.在EOMIP-CCSD方法中,体系电离态的波函数定义为ΨIP=RIPeTΦ0,其中RIP为电离算符:

ri和为电离算符中的r振幅.电离态的能量可由下式得到


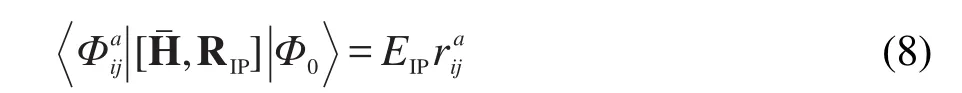
其中Φi,为Φ0所对应的1h,2h1p电离态行列式波函数,EIP是电离能.我们的CC方程包含SOC算符,由于EOM-CC方法的r振幅依赖于CC方法激发算符的t振幅,因此我们所求解的EOMIP方程和CC方程包含相同的SOC算符.考虑旋轨耦合后,r振幅会成为复数,计算过程中同样利用时间反演对称性降低计算量,提高计算效率.24
基态AuX-(X=O,S)电子组态为1σ2π4δ42σ2π*4,12其中1σ、1π和1δ轨道主要是由Au原子的5d轨道和O/S原子的p轨道组合而成,2σ和π*轨道主要是由O/S原子的p轨道和Au原子的5d和6s轨道组合而成.分别从π*、2σ和1δ轨道上电离一个电子即得到AuX 的2Π(1σ2π4δ42σ2π*3),2Σ+(1σ2π4δ42σπ*4)和2Δ(1σ2π4δ32σ2π*4)态.从1π轨道和1σ轨道电离一个电子则得到能量更高的2Π(1σ2π3δ42σ2π*4)和2Σ+(1σπ4δ42σ2π*4)态.如果考虑旋轨耦合效应,2Σ+态变成态,2Π态则会分裂成2Π1/2和2Π3/2,2Δ态分裂成2Δ5/2和2Δ3/2,而且2Σ1/2和2Π1/2,2Π3/2和2Δ3/2由于分别具有相同的对称性还会进一步相互耦合.以闭壳层分子AuX-的基态为参考态,用EOMIP-CC方法即可得到AuX的2Π态、2Σ+态和2Δ态或者2Σ1/2、2Π1/2、2Π3/2、2Δ5/2及2Δ3/2态的能量和性质.本文采用相对论有效势(RECP)方法35,36处理相对论效应.该方法用有效势来代替内层电子对外层电子的作用,并通过适当选取有效势中的参数来计算相对论效应.在计算过程中,Au的内层1s到4f轨道上的60个电子对其它电子的作用使用Stuttgart/Kohn组发展的含旋轨耦合的ECP60MDF有效势37替代,在计算中只考虑5s25p65d106s1电子,所选用的基函数为与相应有效势匹配且考虑了旋轨耦合影响的dhf-QZVPP-2C基函数.38对于O和S原子,使用全电子基组Def2-QZVPPD.39在相关能以及运动方程耦合簇计算中,忽略了Au原子内层5s5p电子,O原子的1s以及S原子的1s2s2p电子对相关能的贡献.不考虑SOC时,这些体系不同对称性下的最低电子态,即能量最低的2Σ+、2Π和2Δ态,可以直接用UCCSD和UCCSD(T)方法进行计算.另外,在不含SOC的计算中,通过比较EOMIP-CCSD和EOMIPCCSDT的计算结果,考察EOMIP-CC计算中三重激发对能量的影响.
本文中所有计算都基于CFOUR程序包,40分别用CCSD、CCSD(T)、SOC-CCSD、SOC-CCSD(T)方法计算了AuO-和AuS-基态1Σ+的键长和频率,用UCCSD、UCCSD(T)、EOMIP-CC方法计算了负离子到AuO和AuS分子2Π、2Σ+和2Δ态所对应的VIP.用EOMIP-SOC-CCSD方法计算了AuO和AuS分子、2Π1/2、2Π3/2、2Δ5/2以及2Δ3/2态的键长和频率,以及这些态所对应的VIP和AIP.
3 计算结果与讨论
分别采用含SOC和不含SOC的CCSD以及CCSD(T)方法计算 AuO-和AuS-基态1Σ+的平衡键长和谐振频率的结果以及一些文献值41,42列在表1中,相应的实验数据以及此前的理论计算结果也列在这个表中.由表1可以看出,对于AuO-,无论是否考虑SOC效应,CCSD得到的键长比CCSD(T)的计算结果要短0.001 nm,而频率要比CCSD(T)的结果大.但是对于AuS-,CCSD和CCSD(T)方法得到的键长和频率基本相同,这说明三重激发对AuS-几乎没有影响.此外,SOC效应使AuO-和AuS-的键长变短0.001-0.002 nm,频率略有增大,这显示闭壳层分子AuO-和AuS-的SOC效应较为显著.SOC使这两个负离子键长变短可能与Au的p1/2轨道参与成键有关.从表1还可以看到,我们用CCSD(T)计算得到的键长要比之前的理论8,12和实验键长8短约0.002-0.003 nm.这应该是由于我们的基组比Wang等12采用的基组大.基组越大,得到的键长通常会越短.
基于不含SOC的CCSD方法下AuO-和AuS-的构型,用UCCSD、UCCSD(T)和EOMIP-CC方法得到的不含SOC情况下的AuO-和AuS-的垂直电离能结果列在表2中.UCCSD和UCCSD(T)对于AuS的2Δ态无法得到收敛的结果.EOMIP-CCSDT对于能量更高的2Π态以及AuS的2Δ态也无法收敛.对比表2中UCCSD和UCCSD(T)的结果可以看出,三重激发使这些态对应的电离能增大约0.2-0.3 eV,说明三重激发对开壳层分子能量的有一定的影响.与EOMIP-CCSDT的结果相比,EOMIP-CCSD高估了这些态对应的电离能,这是因为EOMIP-CCSD方法通常对参考态的描述要优于电离态,因而得到的电离能偏大.对于AuO的2Σ+态以及AuS的2Π和2Σ+态,EOMIP-CCSD和EOMIP-CCSDT的能差在0.1 eV以内,这两个方法对于其它电子态的能差在0.2-0.4 eV之间.比较这两种方法得到的AuO和AuS各电子态的单电离振幅(ri),发现两种方法得到的能差越大,ri振幅差别也越大,比如AuO的2Σ+态,两种方法得到的电离能差异不到0.01 eV,EOMIP-CCSD和EOMIP-CCSDT得到的最大ri振幅分别是0.688和0.672,差异很小;而对于AuO的第二个2Σ+态,EOMIPCCSD和EOMIP-CCSDT得到的最大ri振幅分别是0.657和0.488,考虑三重激发使对应的电离能降低了0.425 eV.AuS的情况与AuO类似.另一方面,AuO和AuS的第二个2Σ+态的双激发振幅raij的模比起别的电子态更大,显示这个态双激发成分更大,因而EOMIP-CCSD误差变大.以上结果表明对于这两个体系的较低电子态2Π态和2Σ+态,EOMIP-CCSD能给出合理的结果,更高电子态则误差增大.
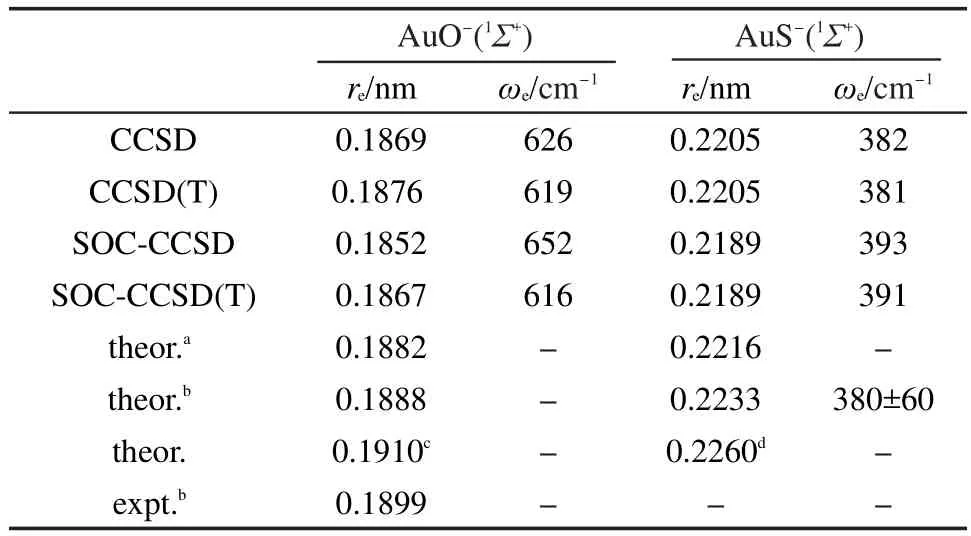
表1 AuO-和AuS-基态的键长(r e)和频率(ωe)Table 1 Bond lengths(r e)and frequencies(ωe)for ground states ofAuO-and AuS-
通常认为对于开壳层体系每个对称性下的最低电子态,UCCSD(T)能给出较精确的计算结果.但是本文中采用UCCSD(T)得到的电离能除了2Δ态外比EOMIP-CCSDT普遍高0.1-0.15 eV左右.由于电离能是两个电子态的能量差,电离能的精度取决于计算这两个态时的误差.CC方法的精度与T1振幅的模有密切联系,一般来说T1振幅的模越小,CC方法的精度越高.我们的计算结果显示,AuO-基态在CCSD水平下T1振幅的模为0.13,而在UCCSD水平下 AuO 的2Π态、2Σ+态和2Δ态 T1振幅的模分别为0.19、0.19和0.12,这说明CC方法对AuO-的基态以及AuO的2Δ态有较高精度,而对AuO的2Π态和2Σ+态则误差较大.这与UCCSD(T)得到的2Δ态对应的电离能与EOMIP-CCSDT得到的结果吻合较好,而2Π和2Σ+态对应的电离能误差较大一致.另外,对比UCCSD和UCCSD(T)结果也显示三重激发对2Δ态所对应的电离能影响小于对2Π和2Σ+态的影响.AuS的2Π态和2Σ+态T1振幅的模更小,所以UCCSD(T)得到的相应电离能比EOMIP-CCSDT高估了不到0.1 eV.另一方面,UCCSD(T)下2Δ态的自旋污染可以忽略,S2约为0.75,而2Π态和2Σ+态的S2分别为0.77和0.80,这也显示UCCSD(T)方法对2Δ态的精度高于2Π态和2Σ+态.这些结果表明EOMIP-CCSDT的结果要比UCCSD(T)可靠.由于CCSDT的计算量为N8,因此本文没有进一步用CCSDT和EOMIP-CCSDT计算这些态的构型和频率.
不含SOC的EOMIP-CCSD计算AuO和AuS的低电子态键长和频率的结果列在表3中,用EOMIPSOC-CCSD方法计算含SOC的AuO和AuS的、2Π1/2、2Π3/2、2Δ5/2及2Δ3/2态的键长和频率结果列在表 4中.比较表1和表3中结果可以看出,对于AuO-和AuS-,从π*轨道电离一个电子,键长变短,频率不变或略有增大.而从2σ轨道电离一个电子,键长变化不大,显示2σ轨道是一个弱成键轨道.虽然1δ是非键轨道,但是从1δ电离一个电子后键长显著增大.对于更高的电子态2Π和2Σ+态,体系键长变长更多,频率变小,这是由于从成键轨道1π和1σ上电离一个电子会削弱化学键所导致的.比较表4和表3中的结果可以看出,对于AuO,2Π3/2态的键长比2Π态短0.0027 nm,而频率增大约30 cm-1,2Π1/2态的键长比2Π态略短而频率也略有增大.与不含SOC的2Σ+态相比,旋轨耦合效应使态的键长减小了0.0014 nm,频率变大 20 cm-1.2Δ5/2态的键长比2Δ态短0.0023 nm,频率变大约30 cm-1.旋轨耦合效应对这些态的键长的影响与对AuO-的键长影响相当.从EOMIP-SOC-CCSD得到的ri振幅可以看到,2Π1/2态波函数中不明显包含态,态中所包含的2Π1/2态的成分约为5%,这说明这两个态之间的耦合较弱.对于表4和表5中列出的2Δ3/2态和第二个2Π3/2态,以及第二个态和2Π1/2态,EOMIP-SOC-CCSD方法的ri振幅显示它们的波函数彼此包含了约45%的组分,也就是说这几个能量较高的电子态考虑SOC效应后混合强烈,用分子轴向总角动量J=3/2,1/2来表示这些分裂电子态更加合理.AuS与AuO的情况类似,不在此详述.另一方面,文献中利用含SOC的MRCI方法计算的结果以及已有实验数据也列在表4中.EOMIP-SOC-CCSD得到的AuO2Π1/2和2Π3/2态的键长比以前MRCI方法得到的键长10短约0.002 nm,频率比之前的结果高大约30 cm-1.从表4中可以看出,本文得到的AuO和AuS含SOC的各电子态的频率与2008年的光电子能谱实验值12吻合得非常好.这表明我们的EOMIP-SOC-CCSD方法能够可靠地描述这两个体系含SOC低电子态的结构和频率.
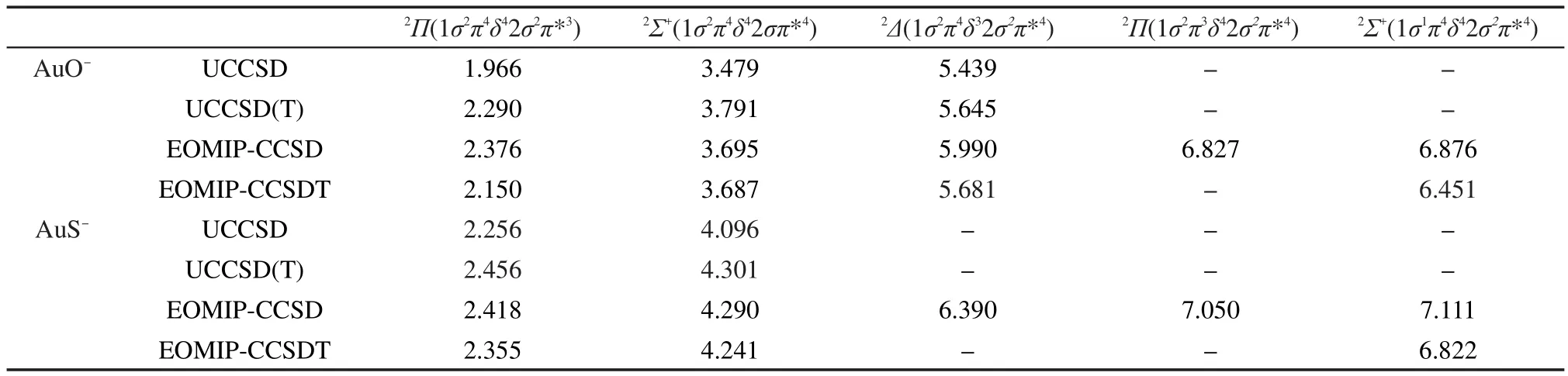
表2 不考虑旋轨耦合时AuO-和AuS-的垂直电离能(单位:eV)Table 2 Vertical ionization energies(unit in eV)ofAuO-and AuS-without spin-orbit coupling
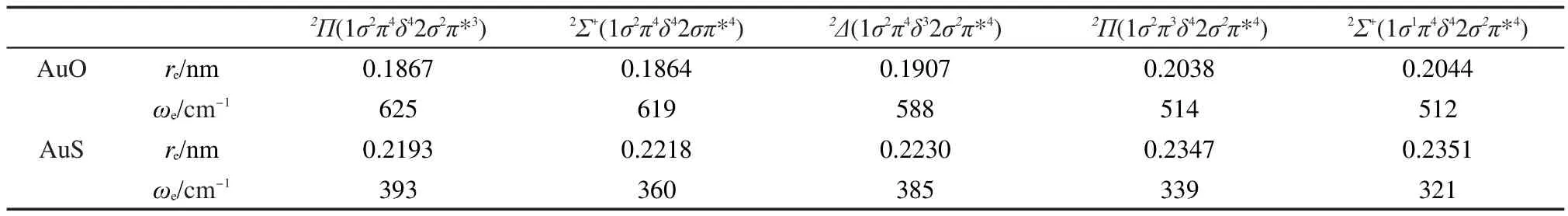
表3 不考虑SOC时AuO和AuS的键长和频率Table 3 Bond lengths and frequencies ofAuOand AuSwithout spin-orbit coupling
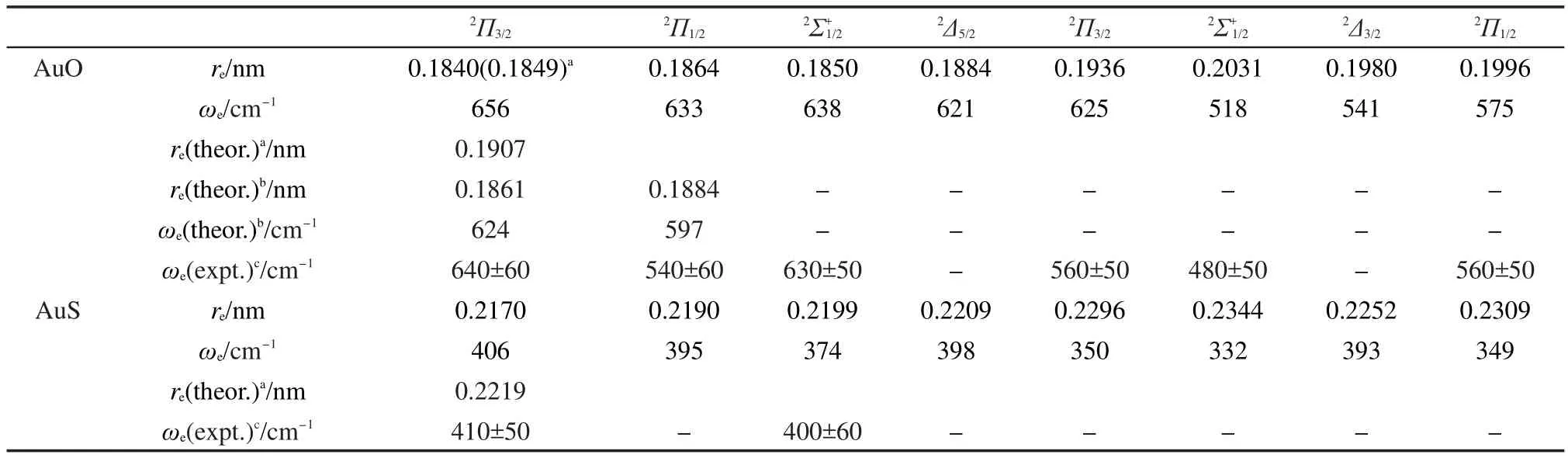
表4 考虑SOC时AuO和AuS的键长和频率Table 4 Bond lengths and frequencies of AuOand AuSwith spin-orbit coupling
本文采用EOMIP-SOC-CCSD方法得到的AuO-和AuS-的垂直电离能和绝热电离能结果列在表5中,其中也包括了以前的理论10,12与实验结果,12EOMIP-SOC-CCSD方法垂直电离能的计算基于表1中1Σ+态SOC-CCSD优化得到的构型.由于表1中SOC-CCSD得到的键长比Wang等12的计算结果短0.003 nm,为了考察键长变化造成的影响,我们基于Wang等计算的键长再次用我们的方法计算了VIP.利用EOMIP-SOC-CCSD方法分别基于我们的键长与Wang的键长得到的VIP差异在0.1 eV以内,可以看到键长差异对电离能的影响可以忽略.结合表4和表5可以看出,对于2Π3/2、2Π1/2及2Δ5/2态由于最大的键长差异不到0.004 nm,因而这些态所对应的垂直电离能和绝热电离能基本相同.能量更高的2Π3/2、、2Δ3/2和2Π1/2态,由于键长增加超过了0.01 nm,导致对应的绝热电离能比垂直电离能小约0.1-0.2 eV.从表5中还可以看到AuO和AuS的2Π1/2态分别比2Π3/2态的能量高0.206和0.129 eV,即AuO的2Π态能量分裂要比AuS更加明显.对于态,AuO和AuS的垂直电离能要比2Σ+态分别高0.062和0.013eV.由于2Π1/2和态之间的耦合会导致2Π1/2能量降低而态能量升高,这说明AuO中2Π1/2和态之间的耦合比AuS这两个态之间的耦合略为显著.这与我们用EOMIP-SOC-CCSD得到AuS的态波函数包含的2Π1/2态成分小于AuO中所混合成分吻合.表2中EOMIP-CCSD得到的AuO和AuS能量最低的2Π态和2Σ+态的能量差分别为1.32和1.87 eV.能差越小,旋轨耦合越显著,这也同我们的结论一致.对于能量更高的2Δ态,第二个2Π态和2Σ+态,一方面由于这些电子态占据轨道包含较多的Au原子的5d轨道成分,虽然SOC对d轨道形状影响较小,但是由于d轨道角动量大,SOC仍然使d轨道的能量分裂显著.另一方面,从表2中可以看到,第二个2Π态和2Σ+态的能量差很小,导致2Δ3/2态和2Π3/2态,2Π1/2态和态之间的耦合强烈.

表5 考虑SOC时AuO-和AuS-的电离能(单位:eV)Table 5 Ionization energies ofAuO-and AuS-with spin-orbit coupling(unit in eV)
我们计算的VIP与2008年的实验结果12相比,对于AuO2Π态的旋轨耦合分裂态2Π3/2和2Π1/2态,误差在0.1 eV以内、2Δ5/2和第二个2Π3/2态的误差大约在0.2 eV.对于能量更高的第二个和2Π1/2态,误差甚至达到了约1.7 eV,这两个电子态的双激发振幅显示,它们的波函数中双激发成分达到了10%以上,所以用EOMIP-SOC-CCSD方法计算这两个电子态的误差很大.将我们的AuO计算结果与Wang等12的理论结果相比较,可以看出,我们得到2Π3/2和2Π1/2态对应的电离能比他们的结果分别大0.02和0.08 eV,而且与实验值吻合更好.对于AuO2Δ态的能量分裂态2Δ5/2和第二个2Π3/2态,其对应的电离能与Wang的结果几乎完全相同.对于能量更高的电子态第二个、2Δ3/2和2Π1/2态,由于我们的方法本身会高估电离态的能量,导致这些态对应的电离能比实验值高出更多.而且这些电离态的双激发成分也较大,如果考虑三重激发的校正,结果应该会更接近实验值.当然,考虑三重激发后也会使低电子态2Π3/2和2Π1/2态对应的电离能下降,这些结果又会偏离实验值更远.对于AuS的低电子态,对比表5中我们得到的VIP与以前的理论和实验数值可以看出,我们低估了2Π3/2和2Π1/2态能量大约0.1 eV左右,高估了,2Δ5/2和第一个2Π3/2态的能量,与实验值的最大误差大约是0.2 eV.与AuO类似,对于能量更高的电子态的误差会更大.
对于能量最低的2Σ+电子态,不考虑SOC效应时EOMIP-CCSD结果与EOMIP-CCSDT吻合非常好.考虑SOC效应后,我们得到的AuS中态能量与Wang等的结果12吻合很好,但是AuO的态能量则比Wang等的结果12低0.2 eV,而Wang等的结果与实验结果吻合非常好.这可能与态中所包含的电子态有关.我们的结果表明态中几乎不包含态的成分,而Wang的结果则显示AuO的态中包含的态成分约为38%,AuS中仅为10%.4Σ-是由组态(πx*)α(π*y)α(σ*)β,(πx*)α(π*y)β(σ*)α和(π*x)β(π*y)α(σ*)α组成,从AuX-出发需要从一个π*轨道上电离一个电子的同时把另一π*轨道上的电子激发到σ*轨道上才能得到,因此EOMIP-CCSD会高估4Σ-态的能量,从而低估4Σ-态和2Σ+态之间的混合.但是2Σ+态与4Σ-态的主要电子组态相差两个轨道,而常用的SOC都是单电子算符或者在平均场近似下的单电子算符,这意味着如果把SOC做微扰处理,在一阶微扰下这两个电子态之间不会相互混合.我们用UCCSD(T)得到的不含旋轨耦合时AuO和AuS的4Σ-态能量比2Σ+态分别低0.1和0.35 eV,可见AuO中4Σ-态与2Σ+态能量接近简并,因此虽然这两个态之间的SOC为二阶小量,它们之间仍然可能有明显的混合.
综上所述,对于AuO和AuS的低电子态,含SOC的EOMIP-CCSD方法能给出可靠的结构和振动频率.在能量计算方面,对于能量较低的电子态,即2Π3/2、2Π1/2、、2Δ5/2以及第二个2Π3/2态,我们的方法可以给出与实验值差别在0.2 eV左右的能量.由于我们的方法本身会高估电离能,对于能量更高的激发态误差会更大,而且这些高激发态双激发明显,需要考虑三重激发的贡献.此外,我们通过比较含SOC和不含SOC的EOMIP-CCSD的结果分析了低电子态的旋轨耦合效应.考虑旋轨耦合效应后,AuX-和AuX的低电子态键长几乎都有不同程度的变短,频率增大.能量最低的2Π1/2和态之间耦合不大,而2Δ3/2态和第二个2Π3/2态,以及第二个态和2Π1/2态之间的耦合严重.以上结果表明我们所发展的EOMIP-SOC-CCSD方法可以合理地描述含SOC开壳层分子AuO和AuS的低电子态,而且此单参考态方法作为一种“黑箱”方法,比CASSCF等多参考态方法使用起来简单.
4 结论
研究了AuO和AuS的低电子态2Π、2Σ+、2Δ态,以及第二个2Π态和2Σ+态的能量和性质.考虑旋轨耦合后,2Π态会分裂为2Π3/2和2Π1/2态,2Σ+态会变成态,2Δ态会分裂为2Δ5/2和2Δ3/2态,具有相同对称性的2Π1/2态和态,2Π3/2态和2Δ3/2态会进一步相互耦合.本工作中采用我们近期发展的含旋轨耦合的EOMIPCCSD方法研究了AuO和AuS低电子态的键长和频率,以及AuO-和AuS-到这些低电子态所对应的垂直电离能和绝热电离能.在此前的工作中,对于含旋轨耦合的开壳层分子低电子态,通常需要采用多参考态方法进行计算,而本文所采用的EOMIPCCSD方法是单参考态方法,使用比多参考态方法更加简单.在不考虑旋轨耦合情况下通过比较EOMIP-CCSD和EOMIP-CCSDT的结果,考察了EOMIP-CC方法中三重激发对能量的影响,结果显示我们的EOMIP-CCSD方法在一定程度上高估了电离能,双激发成分越多,误差越大.此外,通过比较UCCSD(T)和EOMIP-CCSDT的结果,发现通常精度较高的UCCSD(T)方法在处理开壳层体系时,如果T1的模较大并且存在较明显的自旋污染时,得到的结果误差约为0.1-0.15 eV.通过比较含旋轨耦合以及不含旋轨耦合的计算结果,发现考虑旋轨耦合效应后闭壳层分子和开壳层低电子态的键长会在不同程度上变小,频率变大,基态2Π态的分裂态2Π1/2态和态的耦合较弱,但是2Δ态能级分裂后的2Δ3/2态和第二个2Π3/2态,以及第二个态和2Π1/2态之间的耦合显著.我们的方法得到的这些低电子态的结构和振动频率与实验结果吻合较好.虽然EOMIP-SOC-CCSD高估了能量更高的电离态2Δ3/2态、第二个态和2Π1/2态的能量,但是对于其它更低电子态所对应的电离能与已有实验值误差在0.2 eV左右.这显示含旋轨耦合的EOMIP-CCSD方法对于这类原本需要用多参考态方法才能处理的电子态可以得到合理的计算结果.
(1)Pitzer,K.S.Accounts Chem.Res.1979,12,272.
(2)Pyykko,P.;Desclaux,J.P.Accounts Chem.Res.1979,12,276.doi:10.1021/ar50140a002
(4)Love,J.C.;Estroff,L.A.;Kriebel,J.K.;Nuzzo,R.G.;Whitesides,G.M.Chem.Rev.2005,105,1103.doi:10.1021/cr0300789
(5)Griffiths,M.J.;Barrow,R.F.J.Chem.Soc.,Faraday Trans.1977,73,943.doi:10.1039/f29777300943
(6)Citra,A.;Andrews,L.Theochem 1999,489,95.doi:10.1016/S0166-1280(98)00516-8
(7)Sun,Q.;Jena,P.;Kim,Y.D.;Fischer,M.;Gantefor,G.J.Chem.Phys.2004,120,6510.doi:10.1063/1.1666009
(8)Ichino,T.;Gianola,A.J.;Andrews,D.H.;Lineberger,W.C.J.Phys.Chem.A 2004,108,11307.doi:10.1021/jp045791w
(9)O'Brien,L.C.;Hardimon,S.C.;O'Brien,J.J.J.Phys.Chem.A 2004,108,11302.doi:10.1021/jp045812m
(10)O′Brien,L.C.;Oberlink,A.E.;Roos,B.O.J.Phys.Chem.A 2006,110,11954.doi:10.1021/jp063394a
(11)Okabayashi,T.;Koto,F.;Tsukamoto,K.;Yamazaki,E.;Tanimoto,M.Chem.Phys.Lett.2005,403,223.doi:10.1016/j.cplett.2005.01.003
(12)Zhai,H.J.;Bürgel,C.;Bonacic-Koutecky,V.;Wang,L.S.J.Am.Chem.Soc.2008,130,9156.doi:10.1021/ja802408b
(13)Legge,F.S.;Nyberg,G.L.;Peel,J.B.J.Phys.Chem.A 2001,105,790.
(14)Wu,Z.J.J.Phys.Chem.A 2005,109,5951.doi:10.1021/jp0500283
(15)Yao,C.;Guan,W.;Song,P.;Su,Z.M.;Feng,J.D.;Yan,L.K.;Wu,Z.J.Theor.Chem.Acc.2007,117,115.
(16)Becke,A.D.Phys.Rev.A 1988,38,3098.doi:10.1103/PhysRevA.38.3098
(17)Perdew,J.P.Phys.Rev.B 1986,33,8822.doi:10.1103/PhysRevB.33.8822
(18)Lee,C.;Yang,W.;Parr,R.G.Phys.Rev.B 1988,37,785.doi:10.1103/PhysRevB.37.785
(19)Roos,B.O.;Malmqvist,P.Å.Phys.Chem.Chem.Phys.2004,6,2919.doi:10.1039/b401472n
(20)Li,Z.;Suo,B.;Zhang,Y.;Xiao,Y.;Liu,W.Mol.Phys.2013,111,3741.doi:10.1080/00268976.2013.785611
(21)Krylov,A.I.Annu.Rev.Phys.Chem.2008,59,433.doi:10.1146/annurev.physchem.59.032607.093602
(22)Stanton,J.F.;Gauss,J.J.Chem.Phys.1994,101,8938.doi:10.1063/1.468022
(23)Nooijen,M.;Bartlett,R.J.J.Chem.Phys.1995,102,3629.doi:10.1063/1.468592
(24)Tu,Z.Y.;Wang,F.;Li,X.Y.J.Chem.Phys.2012,136,174102.doi:10.1063/1.4704894
(25)Wang,F.;Gauss,J.;van Wüllen,C.J.Chem.Phys.2008,129,064113.doi:10.1063/1.2968136
(26)Kim,I.;Park,Y.C.;Kim,H.;Lee,Y.S.Chem.Phys.2012,395,115.doi:10.1016/j.chemphys.2011.05.002
(27)Cao,Z.L.;Wang,Z.F.;Yang,M.L.;Wang,F.Acta Phys.-Chim.Sin.2014,30(3),431.[曹战利,王治钒,杨明理,王 繁.物理化学学报,2014,30(3),431.].doi:10.3866/PKU.WHXB201401023
(28)Liang,Y.N.;Wang,F.;Li,X.Y.Phys.Chem.Chem.Phys.2013,15,17929.doi:10.1039/c3cp52192c
(29)Stanton,J.F.;Gauss,J.J.Chem.Phys.1999,111,8785.doi:10.1063/1.479673
(30)Manohar,P.U.;Stanton,J.F.;Krylov,A.I.J.Chem.Phys.2009,131,114112.doi:10.1063/1.3231133
(31)Purvis,G.D.,III;Bartlett,R.J.J.Chem.Phys.1982,76,1910.
(32)Wang,F.;Gauss,J.J.Chem.Phys.2008,129,174110.doi:10.1063/1.3000010
(33)Wang,F.;Gauss,J.J.Chem.Phys.2009,131,164113.doi:10.1063/1.3245954
(34)Stanton,J.F.;Bartlett,R.J.J.Chem.Phys.1993,98,7029.doi:10.1063/1.464746
(35)Dolg,M.;Cao,X.Chem.Rev.2011,112,403.
(36)Schwerdtfeger,P.ChemPhysChem 2011,12,3143.doi:10.1002/cphc.201100387
(37)Figgen,D.;Rauhut,G.;Dolg,M.;Stoll,H.Chem.Phys.Lett.2005,311,227.
(38)Weigend,F.;Baldes,A.J.Chem.Phys.2010,133,174102.doi:10.1063/1.3495681
(39)Rappoport,D.;Furche,F.J.Chem.Phys.2010,133,134105.doi:10.1063/1.3484283
(40)Stanton,J.F.;Gauss,J.;Harding,M.E.;Szalay,P.G.;Auer,A.A.;Bartlett,R.J.;Benedikt,U.;Berger,C.;Bernholdt,D.E.;Bomble,Y.J.;Cheng,L.;Christiansen,O.;Heckert,M.;Heun,O.;Huber,C.;Jagau,T.C.;Jonsson,D.;Jusélius,J.;Klein,K.;Lauderdale,W.J.;Matthews,D.A.;Metzroth,T.;Mück,L.A.;O’Neill,D.P.;Price,D.R.;Prochnow,E.;Puzzarini,C.;Ruud,K.;Schiffmann,F.;Schwalbach,W.;Stopkowicz,S.;Tajti,A.;Vázquez,J.;Wang,F.;Watts,J.D.and the integral packages MOLECULE(Almlöf,J.;Taylor,P.R.),PROPS(Taylor,P.R.),ABACUS(Helgaker,T.;Jensen,H.J.A.;Jørgensen,P.;Olsen,J.),and ECP routines by Mitin,A.V.and van Wüllen,C.,CFOUR,Version 1.2;see http://www.cfour.de
(41)Liu,W.J.;van Wüllen,C.J.Chem.Phys.1999,110,3730.doi:10.1063/1.478237
(42)Seminario,J.M.;Zacarias,A.G.;Tour,J.M.J.Am.Chem.Soc.1999,121,411.doi:10.1021/ja982234c
猜你喜欢
数学物理学报(2022年2期)2022-04-26
空间科学学报(2020年6期)2020-07-21
空间科学学报(2020年6期)2020-01-08
环球时报(2019-12-05)2019-12-05
电子制作(2019年16期)2019-09-27
青岛大学学报(工程技术版)(2019年2期)2019-09-10
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
中国惯性技术学报(2015年1期)2015-12-19
航空学报(2015年4期)2015-05-07
