
以生物合成为基础的红霉素A的产量提高和结构改造
2015-03-08 06:28陈单丹吴杰群刘文
生物工程学报 2015年6期
陈单丹,吴杰群,刘文
红霉素是一类广谱大环内酯类抗生素,具有良好的抗革兰氏阳性菌活性和较低的肠胃道毒副作用[1]。自 1952年首次从红色糖多孢菌Saccharopolyspora erythraea (最初命名为红霉素链霉菌 Streptomyces erythreus)[2]中分离获得后,红霉素的使用率一直位于全球抗生素类药物的前列。从化学结构上看,红霉素由红霉内酯 (Erythronolid)、L-碳霉糖 (L-mycarose) 和D-脱氧糖胺 (D-desosamine) 3部分以O-糖苷键连接而成。红霉素A作为活性最好的天然组分,区别于红霉素B、C、D的特征在于大环骨架的羟化和糖基单元的甲基化程度不同[3](图1)。以红霉素A为代表的第一代红霉素在酸性条件下容易脱水缩合形成螺缩酮,继而丧失生物活性[4]。此外,随着红霉素在临床上的大量使用,越来越多的耐药性问题也引起了人们的关注[5]。通过红霉素A的结构修饰,不仅能够提高化合物对酸的稳定性,克服细菌耐药性的问题,甚至对抗菌谱也有较大的扩展[6]。以阿齐霉素(Azithromycin)[7]、克拉霉素 (Clarithromycin)[8]、罗红霉素 (Roxithromycin)[9]为代表的第二代红霉素,和以泰利霉素 (Telithromycin)[10]、赛红霉素 (Cethromycin)[11]为代表的第三代红霉素都是在红霉素A的基础上通过化学结构修饰开发得到的[12](图1),同时也都是当今主流的医用抗生素。由此可见,针对红霉素A的产量提高研究具有重要的经济价值,而对其进行结构改造更具有巨大的新药开发潜力。由于红霉素 A的结构复杂,采用化学方法进行合成和修饰的能力比较有限[13]。在掌握生物合成机制的前提下,人们可以特异性地从遗传水平上操纵红霉素产生菌种的代谢途径,构建基因重组菌株用于大量发酵最为有效的组分红霉素 A;或者利用组合生物合成 (Combinatorial biosynthesis)技术对红霉素A的生物合成途径进行改造,获得结构复杂的红霉素类似物库用于筛选和发展具有应用前景的药物[14]。

图1 红霉素类抗生素的代表性药物[3,7-11]Fig. 1 Representative erythromycins[3,7-11].
近年来,人们对于红霉素A的生物合成机制已经有了全面的研究和深刻的认识。除了红色糖多孢菌以外,红霉素的产生菌种主要是土壤微生物,包括红霉素气微菌 Aeromicrobium erythreum[15]和巴西诺卡菌 Nocardia brasiliensis[16]。基于产生菌种的基因组测序[17],红霉素生物合成基因簇的信息不断获得丰富[18],包括关键蛋白的结构功能鉴定、催化机制推导[19-22]以及生物合成途径的阐明[23]等。值得一提的是,近期在中性嗜盐的红色放线多孢菌 Actinopolyspora erythraea 中也报道了红霉素和新型红霉内酯的产生[24]。本实验室通过对这株嗜盐菌的全基因组测序,定位得到全新的红霉素生物合成基因簇;结合化学型-基因型关联分析,推导了其中红霉素类化合物的生物合成机制[25]。本文综述了近年来基于红霉素A生物合成机制展开的以产量提高和结构改造为目标的相关研究进展。
1 红霉素A的生物合成机制
红霉素A的生物合成可以分为大环内酯骨架6-脱氧红霉内酯B (6-deoxyerythronolide B,6-dEB) 的形成和后修饰两大步骤[23]。
6-dEB 的生物合成由 I型聚酮合酶(Polyketide synthase,PKS) 复合酶系催化。该复合酶系含有 3个酶蛋白亚基:DEBS1、DEBS2和DEBS3,总共包括1个起始模块 (Module) 和6个延伸模块,涉及3个基本功能域 (Domain):β-酮基硫酯合成酶 (Ketosynthase,KS)、酰基转移酶 (Acyltransferase,AT)、酰基载体蛋白(Acyl carrier protein,ACP)[26]。起始模块中的AT特异性识别丙酰辅酶A (Coenzyme A,CoA),将其转移到ACP上作为起始单元。起始单元随后被转移到第一个延伸模块的KS上,同时第一个延伸模块中的 AT特异识别甲基丙二酰 CoA并将其转移到第一个延伸模块的 ACP上,KS催化起始单元丙酰基与延伸单元甲基丙二酰基缩合形成 β-酮完成第一轮延伸。其他 5个延伸模块采用类似的机理依次线性完成缩合,连接在PKS上的聚酮链在每一轮延伸后都会增加两碳单元。此外,延伸模块还选择性含有 β-酮基还 原 酶 (β-ketoreductase, KR)、 脱 水 酶(Dehydratase, DH)、 烯 醇 还 原 酶(Enoylreductase,ER) 功能域,分别特异性地将延伸聚酮链的 β-羰基还原至羟基、反式烯基或完全饱和的状态,最终形成一个十四碳的聚酮链。在末端硫酯酶 (Thioesterase,TE) 的催化下,该聚酮中间体从PKS上解离下来,环化形成 6-dEB[27](图 2)。
大环内酯骨架 6-dEB在形成之后,还需要经历一系列后修饰反应才能获得成熟的终产物红霉素A,以及包括红霉素B、C、D在内的中间产物。首先,细胞色素P450氧化酶EryF在6-dEB的 C6位进行羟化,形成红霉内酯 B(Erythronolide B,EB)[28]。然后,糖基转移酶EryBV在EB的C3位羟基上连接L-碳霉糖,形成 3-O-碳霉糖基红霉内酯 B (3-α-mycarosyl erythronolide B,MEB)。紧接着,另一个糖基转移酶 EryCⅢ在MEB的 C5位羟基上连接D-脱氧糖胺,形成首个具有生物活性的中间体——红霉素D[29]。红霉素D在细胞色素P450氧化酶EryK的催化下完成C12位的羟化,合成红霉素C;再在 S-腺苷甲硫氨酸 (S-adenosyl methionine,SAM) 依赖的甲基化酶 EryG作用下完成碳霉糖单元 C3’’位的羟甲基化,合成最终产物红霉素A。此外,红霉素D也可以以较低的效率先在EryG的作用下完成碳霉糖单元的羟甲基化,得到红霉素B;再通过EryK的作用,合成红霉素A[30](图2)。作为后修饰不完全的中间体,红霉素B、C、D等组分在原始产生菌种的发酵液中广泛存在、难以去除,被视为副产物。
2 以生物合成为基础的红霉素A的产量提高研究
由于红霉素A的结构较为复杂,采用化学合成工艺累积步骤繁琐、成本较高,因此,生产上主要采用微生物发酵的方法获得。红霉素发酵产业所面临的主要问题是发酵单位不高、组分比例不佳,这也使我国红霉素A原料药的竞争力受到了很大的限制。

图2 红霉素A的生物合成途径[23]Fig. 2 Biosynthetic pathway of erythromycin A[23].
针对发酵单位不高的问题,除了传统的诱变育种以外,人们也基于红霉素A的生物合成机制开发了若干行之有效的高产菌种遗传改造策略。例如,每分子红霉素A的生物合成都需要引入1分子的丙酰CoA和6分子的甲基丙二酰 CoA。因此,提高这两种前体特别是甲基丙二酰CoA的体内供给量将极大地提高红霉素的发酵产量。Reeves等[31-32]通过过量表达甲基丙二酰CoA变位酶 (Methylmalonyl-CoA mutase,MCM),强化了甲基丙二酰 CoA前体的体内供给量,获得了红霉素的高产菌株。如果把红色糖多孢菌看成红霉素生物合成的车间,那么最有效直接的提高生产速度的方法不是改变生产过程中某一个环节的工艺,而是重新建设一条新的生产线。本实验室在红色糖多孢菌体内倍增了红霉素的PKS编码基因,使红霉素的产量提高了近50%,并使发酵周期缩短了1/3。此外,由于大肠杆菌 Escherichia coli具有生长代谢快速、基因操作方便、遗传背景清楚等优点,是理想的红霉素A异源表达宿主。Pfeifer课题组通过导入红霉素生物合成基因簇中的全部结构基因,实现了红霉素A在大肠杆菌中的异源表达[33]。他们还通过强化外源蛋白的表达,在大肠杆菌中合理地调控丙酰CoA和甲基丙二酰CoA前体的生物合成,提高了红霉素的产量[34]。
针对组分比例不佳的问题,解决方法在于选育红霉素A的绝对优势菌株。根据红霉素A的骨架结构后修饰途径,控制红霉素从D组分到A组分的转化需要两个关键的后修饰蛋白参与:细胞色素P450氧化酶EryK和SAM依赖的甲基化酶 EryG。这两个酶催化效力的不足直接导致了红霉素B和C的积累[30]。本实验室首先通过同源重组的方式把2个拷贝的eryK和1个拷贝的eryG导入到红色糖多孢菌E3染色体中。当羟化酶基因 eryK与甲基化酶基因 eryG的拷贝数为3∶2时,杂质红霉素B和C完全消失,红霉素A的产量也提高了约30%[35]。这是首例采用基因工程的方法从根源上把红霉素杂质组分 B和 C完全除去。为了克服菌种发酵的不稳定性,保证高产菌株在没有抗性压力的情况下依然维持稳定的基因型和高产表型,本实验室尝试采用由放线菌噬菌体ΦC31整合酶介导的位点特异性重组构建遗传稳定的红霉素A高产菌株。我们在红色糖多孢菌基因组中一段功能丧失的 PKS区域导入基于ΦC31整合酶整合的基因插入盒,将8个拷贝的ΦC31整合酶特异性识别的细菌插入序列(Bacterial attachment site,attB) 导入上述PKS区域,以后便可以实现目的基因的稳定整合 (图3),也为红霉素A的工业生产带来了便利[36]。
3 以生物合成为基础的红霉素A的结构改造研究
红霉素A的生物合成包含两大步骤,相应地,对其进行结构改造的策略也主要从两方向展开:其一是对编码6-dEB形成的PKS基因进行遗传改造,定向改变大环内酯的骨架结构;其二是对红霉素合成的后修饰途径进行遗传改造,特别是定向改变糖基单元的结构。
3.1 大环内酯骨架的结构改造
Ⅰ型PKS的组织结构和催化功能之间存在着一一对应的线性逻辑关系,在蛋白亚基、模块、功能域水平上的任何改变,都会影响大环内酯合成反应中的某一环节,从而引起骨架结构发生相应的变化。由此产生的新型红霉内酯大多能够经历后修饰反应,转化为相应的红霉素类似物。目前,对红霉素大环内酯骨架的结构改造研究主要集中在模块和功能域的改变上,常见的策略包括功能域的替换、失活或插入,以及模块的替换、重组或改造等[37]。

图3 红霉素A高产菌株的构建策略和发酵产物分析[36]Fig. 3 Strategy of constructing high erythromycin A-producing strains and analysis of the fermentation products[36].
3.1.1 Ⅰ型PKS功能域水平上的遗传操作
在Ⅰ型PKS中,AT功能域负责小分子羧酸底物的识别和上载。将其替换为识别不同底物的AT,可以在聚酮链的合成过程中掺入不同的结构单元。在6-dEB的形成过程中,6个延伸模块的AT都特异识别和上载甲基丙二酰CoA。将特异识别丙二酰CoA的AT分别替换模块1到模块6的AT,能够分别产生在大环内酯C12、C10、C8、C6、C4和 C2位缺少甲基的红霉内酯 1-6[38-41]。类似地,采用来自尼达霉素(Niddamycin) 的PKS模块7的识别乙基丙二酰CoA的AT替换红霉素PKS模块5的AT,则可获得C6位乙基取代的红霉内酯7[42](图4)。
Ⅰ型PKS的延伸模块选择性含有KR、DH和ER功能域,通过影响延伸中聚酮链的β-羰基的还原水平,改变大环内酯的骨架结构。对红霉素PKS模块6中的KR和模块4中的ER进行点突变失活,可以分别得到C3位酮基取代的红霉内酯 8和 C6-C7位不饱和的红霉内酯9[43-44]。此外,直接敲除模块6中的KR将影响与之相连的AT的底物识别特异性,除产生化合物8外,还意外地产生了化合物10[37](图5)。而对红霉素PKS唯一的模块4中的DH进行点突变失活,将导致DEBS2功能的完全丧失[45]。

图 4 延伸模块 AT功能域替换所产生的新型红霉内酯1-7[38-42]Fig. 4 Novel erythronolides 1-7 produced by substituting the AT domains in the extender modules[38-42].

图5 延伸模块KR和ER功能域失活所产生的新型红霉内酯8-10[37,43-44]Fig. 5 Novel erythronolides 8-10 produced by inactivating the KR and ER domains in the extender modules[37,43-44].
向红霉素PKS的相关模块插入原本不存在的KR、DH和ER功能域,同样能够产生一些结构类似物。McDaniel等用雷帕霉素(Rapamycin) PKS模块4中的DH-KR区域分别取代红霉素PKS模块2和模块6中的KR,产生大环内酯C10-C11位和C2-C3位含有不饱和烯键的红霉内酯 11和 12。而用雷帕霉素 PKS模块 1中的 DH-ER-KR区域分别取代红霉素PKS模块2和模块5中的KR,则产生大环内酯C11位和C5位脱羟基的红霉内酯13和14。然而,用此区域替换红霉素PKS模块6的KR时,并未发现C3位脱羟基的化合物出现,反而有较多的化合物12和15积累[39]。由此可见,替换后的模块ER无活性,KR只有部分活性。这点可以反映出插入功能域的活性受到蛋白整体结构的影响。许多由Ⅰ型PKS编码的聚酮化合物,如泰乐菌素 (Tylosin)[46]、苦霉素 (Pikromycin)[47]、多杀菌素 (Spinosyn)[48]、竹桃霉素(Oleandomycin)[49]等,其起始模块在AT-ACP区域的前端含有 β-酮酰基合成酶 (β-ketoacyl synthase,KSQ) 功能域。首先,AT识别丙二酰CoA,将其结合到ACP上;随后,KSQ催化丙二酰硫醇脱羧形成乙酰基;最后,再转移到第一个延伸模块的KS上进行后续的合成。把竹桃霉素起始模块中的KSQ与雷帕霉素模块2中识别丙二酰CoA的AT相连,用以替换红霉素PKS起始模块中识别丙酰CoA的AT,该杂合的起始模块最终把乙酰基传递到红霉素PKS第一个延伸模块的 KS上,产生 C13位带有甲基的红霉内酯 16[50](图6)。
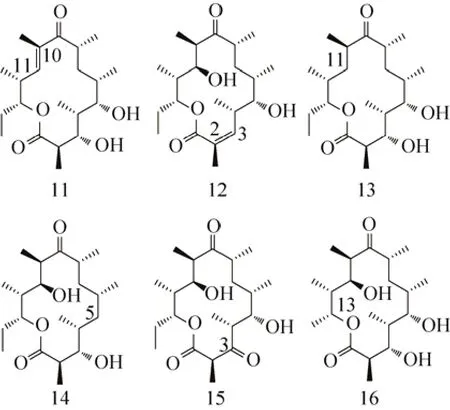
图6 延伸模块KR和ER-KR功能域插入所产生的新型红霉内酯11-16[39,50]Fig. 6 Novel erythronolides 11-16 produced by inserting the KR and ER-KR domains in the extender modules[39,50].
3.1.2 Ⅰ型PKS模块水平上的遗传操作
红霉素和阿维菌素 (Avermectin) PKS的起始模块均只含有AT-ACP结构域。前者的AT能够特异性识别丙酰CoA为起始单元,并将其上载到同模块的ACP上;后者的AT具有较为宽泛的底物选择性,能够识别异丁酰CoA或者2-甲基丁酰CoA为起始单元,将其上载到同模块的ACP上[51]。用阿维菌素PKS的起始模块替换红霉素PKS的起始模块,杂合的PKS能合成大环内酯 C13位上连接异丙基或仲丁基的红霉内酯 17和 18[52](图 7)。

图7 起始模块替换所产生的新型红霉内酯17-18[52]Fig. 7 Novel erythronolides 17-18 produced by substituting the loading module[52].
红霉素DEBS3末端的TE功能域负责聚酮链的水解,并将其环化成为十四元环骨架6-dEB[53]。该TE对聚酮链的长度有较高的容忍度,能够识别和催化不同长短的聚酮链形成不同大小的内酯环。将TE连接到DEBS1的末端能够产生六元环内酯化合物19[54](图8A)。而将含有红霉素PKS模块5和6及TE的DEBS3基因导入到天蓝色链霉菌 Streptomyces coelicolor CH999中,在不含起始模块的情况下,DEBS3仍能识别甲基丙二酰CoA为底物,脱羧形成丙酰基继续往下延伸,最终环化形成六元环内酯20[55](图8B)。这与前面所述起始模块含有KRQ结构域的催化机制十分相似。化合物 19与 20的构型不同,原因在于KR催化的立体选择性:其中,模块 1的 KR在催化β-酮基还原成羟基时,专一地产生R构型产物;而其他模块的KR则都产生 S构型的产物[56-57]。KR属于短链脱氢/还原酶 (Short-chain dehydrogenase/reductase,SDR) 家族[58],催化产生R构型化合物的 KR活性中心一侧含有高度保守的天冬氨酸残基,催化产生S构型化合物的 KR活性中心则缺少此天冬氨酸残基而在活性中心相反的一侧含有色氨酸残基。如此细微差异使聚酮化合物在进入KR活性中心时的方向不同,从而导致还原产物形成不同的立体构型[44,57]。将TE连接到模块5的末端能够产生十二元环内酯21[59](图8C)。将雷帕霉素PKS模块2或模块5插入到红霉素PKS模块1之后,能够产生十六元环化合物22和23以及在C13位环化的十四元环产物 24和 25[60](图 8D)。
近年来对 DEBS的蛋白晶体学研究日益深入,基于对蛋白立体结构和蛋白亚基相互作用的分析,可以人为地对PKS模块进行定向改造。研究发现,ACP功能域的结构对延伸中的聚酮链在模块内和模块间的传递都至关重要[61]。通过改造红霉素PKS模块3中的ACP,将其部分替换成模块2的ACP,可以实现整个模块3的重复使用,形成四酮产物26[62](图9)。
3.2 后修饰途径中糖基单元的结构改造
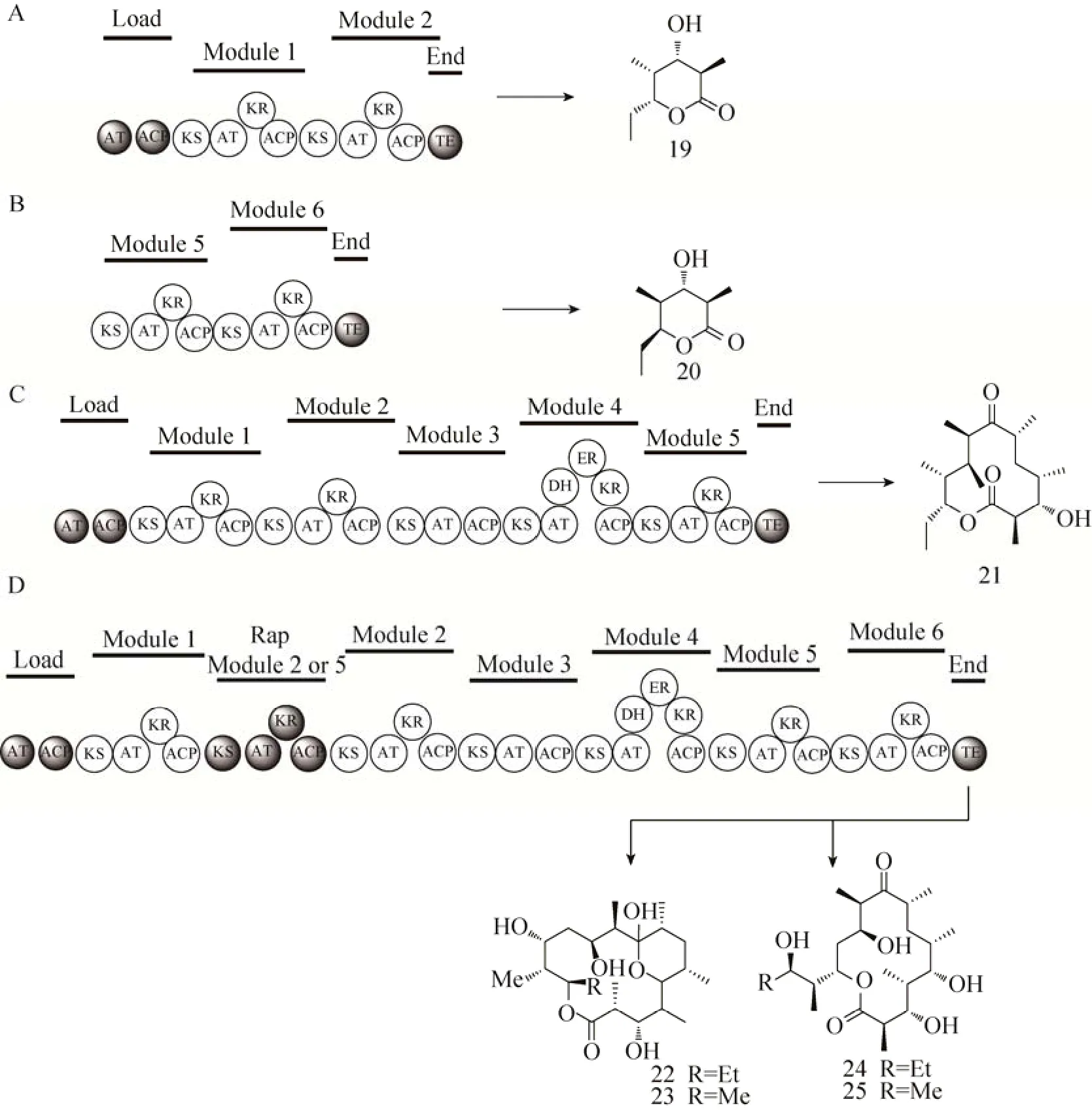
图8 重组模块的结构与其催化合成的产物[54-55,59-60] (A:TE功能域与起始模块、延伸模块1-2组合所产生的新型六元内酯19;B:TE功能域与延伸模块5-6组合所产生的新型六元内酯20;C:TE功能域与起始模块、延伸模块1-5组合所产生的新型十二元内酯21;D:雷帕霉素延伸模块2或5插入所产生的新型十六元内酯22和23、新型十四元内酯24和25)Fig. 8 Domain organization of hybrid PKSs and their products[54-55,59-60]. (A) A novel six-membered erythronolide 19 produced by coupling the TE domain with the loading module and the extender modules 1-2. (B) A novel six-membered erythronolide 20 produced by coupling the TE domain with the loading module and the extender modules 5-6. (C) A
红霉素 A生物合成的后修饰过程包括在6-dEB上的两步羟基化、两步糖基化以及在糖基上的一步甲基化。由于糖基单元能够直接参与天然产物与靶点之间的相互作用,或者参与靶细胞的识别[63],因此,在红霉素A后修饰途径研究中最有价值的部分是大环内酯骨架的糖基化。C5位的D-脱氧糖胺几乎存在于所有的十四元大环内酯类活性天然产物中,被认为是红霉素A与核糖体结合从而抑制细菌生长的一个关键因素[37,64]。而C3位L-碳霉糖的存在似乎对于红霉素A的抗菌活性贡献不大,去除以后的红霉素衍生物反而能够克服一些已经出现的细菌耐药性问题[65]。由此提示,对于红霉素A糖基novel twelve-membered erythronolide 21 produced by coupling the TE domain with the loading module and the extender modules 1-5. (D) Novel sixteen-membered erythronolides 22 and 23 and fourteen-membered erythronolides 24 and 25 produced by inserting the extender module 2 or 5 of rapamycin.单元的结构改造着眼点应该放在由糖基转移酶EryBV催化红霉素内酯环 C3位羟基的糖基化上。体外测活实验证实,EryBV对糖基单元具有比较宽泛的底物选择性,能够识别一系列的糖基作为底物以较低的催化效率合成红霉素类似物 27−37[66]。

图9 改造ACP功能域使模块3重复催化链延伸[62]Fig. 9 Engineered module 3 with the chimeric ACP domain exhibits iterative chain elongation[62].

图10 EryBV底物宽泛性所产生的红霉素中间体[66]Fig. 10 Erythromycin intermediates based on the substrate promiscuity of EryBV[66].

图 11 EryCⅢ底物宽泛性所产生的红霉素类似物[68-69]Fig. 11 Erythromycin analogues based on the substrate promiscuity of EryCⅢ[68-69].
相对于EryBV,糖基转移酶EryCⅢ对糖基单元具有较高的底物特异性[67]。有报道显示,EryCⅢ能够把 D-麦氨糖 (D-mycaminose) 转移到大环内酯C5位的羟基上,从而形成新的红霉素类似物红霉素M[68]。当缺乏D-脱氧糖胺时,EryCⅢ也可能识别 L-碳霉糖,催化形成化合物38[69]。
许多糖基转移酶对底物的容忍性都比较高,可能将不同的糖基转移至红霉素大环内酯骨架。例如,来自泛温链霉菌 Streptomyces eurythermus的 AngMⅡ是泰乐菌素生物合成中糖基转移酶。以泰乐酮 (Tylactone) 作为天然底物,将 D-麦氨糖转移到 C5位的羟基上,产生化合物 39。研究发现,AngMⅡ同样能够识别EB 作为底物,糖基化产生化合物 40[69],并且糖基化的位置是C5位的羟基,而不是C3位的羟基。又如,苦霉素生物合成中负责C5位羟基糖基化的糖基转移酶对底物的容忍性也较高,能够识别一系列 6-dEB类似物,可以合成超过20种以上的新化合物[70]。

图 12 糖基转移酶 AngMⅡ催化产生的新型红霉素40[69]Fig. 12 Novel erythromycin 40 generated by the glycosyltransferase AngMⅡ[69].
4 总结
作为红霉素最为有效的天然组分和化学修饰获得第二代、第三代红霉素药物的底物,红霉素A的产量提高和结构改造具有非常重要的意义和价值。基于生物合成机制,可以明确与红霉素A生物合成直接相关的关键因素,包括PKS前体、催化蛋白等。在高产菌株遗传改造方面,传统的诱变育种技术目标不明确,结果不稳定,弊端显而易见。通过对生物合成中的限速步骤的预测,可以对高产菌株的遗传改造进行良好的指导。例如,在红霉素A产生菌的体内增加催化蛋白和PKS前体的浓度,可以提高红霉素的发酵产量[31-32];对后修饰途径中的关键基因进行表达强化,则可以特异性提高红霉素A的组分比例[35-36]。在化合物结构改造方面,采用化学合成或修饰的方法所能进行的结构衍生也非常有限。通过对关键催化蛋白的结构和功能的分析,则可以更好地利用组合生物合成技术获得大量结构复杂的红霉素 A类似物。目前的研究重点主要集中在 6-dEB大环内酯骨架结构的改造和后修饰步骤中的糖基化反应。我们相信,充分利用生物合成的基本原理进行菌种的遗传改造,可以弥补化学合成和修饰的不足,降低红霉素A的生产成本,促进新药的研发。
值得一提的是,以对天然产物生物合成机制的理性认识指导微生物菌种遗传改造的这种“师从于自然”的理念对于大量具有高附加值的抗生素类药物同样适用。本实验室长期致力于采用化学思想和生物学技术解决在大宗微生物药物发酵工业中的实际问题,在阿维菌素(Avemectin)[71]、硫链丝菌素 (Thiostrepton)[72-73]、他克莫司 (Tacrolimus)[74]、林可霉素(Lincomycin)[75]的产生菌种改造方面做出了大量基础型和应用型的研究工作。在改造菌种的发酵产品中,有效组分的产量提高、比例优化,可以大幅度地提高生产效益,简化后续工艺,增强企业竞争力,同时促进微生物药物的更新升级。
[1] McGuire JM, Bunch RL, Anderson RC, et al.Ilotycin, a new antibiotic. Antibiot Chemother,1952, 2(6): 281−283.
[2] Labeda DP. Transfer of the type strain of Streptomyces-erythraeus (waksman 1923)waksman and henrici 1948 to the genus Saccharopolyspora lacey and goodfellow 1975 as Saccharopolyspora-erythraea sp-nov, and designation of a neotype strain for Streptomyces-erythraeus. Int J Syst Evo Bacteriol,1987, 37(1): 19−22.
[3] Kibwage IO, Hoogmartens J, Roets E, et al.Antibacterial activities of erythromycins A, B, C,and D and some of their derivatives. Antimicrob Agents Chemother, 1985, 28(5): 630−633.
[4] Omura S, Tsuzuki K, Sunazuka T, et al.Macrolides with gastrointestinal motor stimulating activity. J Med Chem, 1987, 30(11): 1941−1943.
[5] Leclercq R. Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications.Clin Infect Dis, 2002, 34(4): 482−492.
[6] Ma ZK, Nemoto PA. Discovery and development of ketolides as a new generation of macrolide antimicrobial agents. Curr Med Chem, 2002, 1(1):15−34.
[7] Bright GM, Nagel AA, Bordner J, et al. Synthesis,in vitro and in vivo activity of novel 9-deoxo-9a-AZA-9a-homoerythromycin A derivatives; a new class of macrolide antibiotics,the azalides. J Antibiot, 1988, 41(8): 1029−1047.
[8] Morimoto S, Takahashi Y, Watanabe Y, et al.Chemical modification of erythromycins. I.synthesis and antibacterial activity of 6-O-methylerythromycins A. J Antibiot, 1984,37(2): 187−189.
[9] Gasc JC, Dambrieres SG, Lutz A, et al. New ether oxime derivatives of erythromycin A. A structure-activity relationship study. J Antibiot,1991, 44(3): 313−330.
[10] Denis A, Agouridas C, Auger JM, et al. Synthesis and antibacterial activity of HMR 3647 a new ketolide highly potent against erythromycinresistant and susceptible pathogens. Bioorg Med Chem Lett, 1999, 9(21): 3075−3080.
[11] Or YS, Clark RF, Wang SY, et al. Design,synthesis, and antimicrobial activity of 6-O-substituted ketolides active against resistant respiratory tract pathogens. J Med Chem, 2000,43(6): 1045−1049.
[12] Bosnar M, Kelnerić Z, Munić V, et al. Cellular uptake and efflux of azithromycin, erythromycin,clarithromycin, telithromycin, and cethromycin.Antimicrob Agents Chemother, 2005, 49(6):2372−2377.
[13] Shen B, Liu W, Nonaka K. Enediyne natural products: biosynthesis and prospect towards engineering novel antitumor agents. Curr Med Chem, 2003, 10(21): 2317−2325.
[14] Walsh CT. Combinatorial biosynthesis of antibiotics: challenges and opportunities.Chembiochem, 2002, 3(2-3): 125−134.
[15] Miller ES, Woese CR, Brenner S. Description of the erythromycin-producing bacterium Arthrobacter sp. strain NRRL B-3381 as Aeromicrobium erythreum gen. nov., sp. nov.. Int J Syst Bacteriol, 1991, 41(3): 363−368.
[16] Mikami Y, Yazawa K, Nemoto A, et al. Production of erythromycin E by pathogenic Nocardia brasiliensis. J Antibiot, 1999, 52(2): 201−202.
[17] Oliynyk M, Samborskyy M, Lester JB, et al.Complete genome sequence of the erythromycinproducing bacterium Saccharopolyspora erythraea NRRL23338. Nat Biotechnol, 2007,25(4): 447−453.
[18] Weber JM, Leung JO, Maine GT, et al.Organization of a cluster of erythromycin genes in Saccharopolyspora erythraea. J Bacteriol, 1990,172(5): 2372−2383.
[19] Savino C, Montemiglio LC, Sciara G, et al.Investigating the structural plasticity of a cytochrome P450: three-dimensional structures of P450 EryK and binding to its physiological substrate. J Biol Chem, 2009, 284(42):29170−29179.
[20] Cupp-Vickery JR, Poulos TL. Structure of cytochrome P450eryF involved in erythromycin biosynthesis. Nat Struct Biol, 1995, 2(2):144−153.
[21] Cortes J, Haydock SF, Roberts GA, et al. An unusually large multifunctional polypeptide in the erythromycin-producing polyketide synthase of Saccharopolyspora erythraea. Nature, 1990,348(6297): 176−178.
[22] Summers RG, Donadio S, Staver MJ, et al.Sequencing and mutagenesis of genes from the erythromycin biosynthetic gene cluster of Saccharopolyspora erythraea that are involved in L-mycarose and D-desosamine production.Microbiology, 1997, 143(10): 3251−3262.
[23] Staunton J, Wilkinson B. Biosynthesis of erythromycin and rapamycin. Chem Rev, 1997,97(7): 2611−2630.
[24] Huang SX, Zhao LX, Tang SK, et al.Erythronolides H and I, new erythromycin congeners from a new halophilic actinomycete Actinopolyspora sp. YIM90600. Org Lett, 2009,11(6): 1353−1356.
[25] Chen DD, Feng JY, Huang L, et al. Identification and characterization of a new erythromycin biosynthetic gene cluster in Actinopolyspora erythraea YIM90600, a novel erythronolideproducing halophilic actinomycete isolated from salt field. PLoS ONE, 2014, 9(9): e108129.
[26] Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep,2001, 18(4): 380−416.
[27] Khosla C, Tang YY, Chen AY, et al. Structure and mechanism of the 6-deoxyerythronolide B synthase. Annu Rev Biochem, 2007, 76: 195−221.[28] Weber JM, Leung JO, Swanson SJ, et al. An erythromycin derivative produced by targeted gene disruption in Saccharopolyspora erythraea.Science, 1991, 252(5002): 114−117.
[29] Salah-Bey K, Doumith M, Michel JM, et al.Targeted gene inactivation for the elucidation of deoxysugar biosynthesis in the erythromycin producer Saccharopolyspora erythraea. Mol Gen Genet, 1998, 257(5): 542−553.
[30] Lambalot RH, Cane DE, Aparicio JJ, et al.Overproduction and characterization of the erythromycin C-12 hydroxylase, EryK.Biochemistry, 1995, 34(6): 1858−1866.
[31] Reeves AR, Brikun IA, Cernota WH, et al.Engineering of the methylmalonyl-CoA metabolite node of Saccharopolyspora erythraea for increased erythromycin production. Metab Eng,2007, 9(3): 293−303.
[32] Reeves AR, Brikun IA, Cernota WH, et al. Effects of methylmalonyl-CoA mutase gene knockouts on erythromycin production in carbohydrate-based and oil-based fermentations of Saccharopolyspora erythraea. J Ind Microbiol Biotechnol, 2006,33(7): 600−609.
[33] Zhang HR, Wang Y, Wu JQ, et al. Complete biosynthesis of erythromycin A and designed analogs using E. coli as a heterologous host. Chem Biol, 2010, 17(11): 1232−1240.
[34] Jiang M, Pfeifer BA. Metabolic and pathway engineering to influence native and altered erythromycin production through E. coli. Metab Eng, 2013, 19: 42−49.
[35] Chen Y, Deng W, Wu JQ, et al. Genetic modulation of the overexpression of tailoring genes eryK and eryG leading to the improvement of erythromycin a purity and production in Saccharopolyspora erythraea fermentation. Appl Environ Microbiol, 2008, 74(6): 1820−1828.
[36] Wu JQ, Zhang QL, Deng W, et al. Toward improvement of erythromycin A production in an industrial Saccharopolyspora erythraea strain via facilitation of genetic manipulation with an artificial attB site for specific recombination. Appl Environ Microbiol, 2011, 77(21): 7508−7516.
[37] McDaniel R, Welch M, Hutchinson CR. Genetic approaches to polyketide antibiotics. Chem Rev,2005, 105(2): 543−558.
[38] Ruan X, Pereda A, Stassi DL, et al.Acyltransferase domain substitutions in erythromycin polyketide synthase yield novel erythromycin derivatives. J Bacteriol, 1997,179(20): 6416−6425.
[39] McDaniel R, Thamchaipenet A, Gustafsson C, et al. Multiple genetic modifications of the erythromycin polyketide synthase to produce a library of novel "unnatural" natural products. Proc Natl Acad Sci USA, 1999, 96(5): 1846−1851.
[40] Liu L, Thamchaipenet A, Fu H, et al. Biosynthesis of 2-Nor-6-deoxyerythronolide B by rationally designed domain substitution. J Am Chem Soc,1997, 119(43): 10553−10554.
[41] Petkovic H, Lill RE, Sheridan RM, et al. A novel erythromycin, 6-desmethyl erythromycin D, made by substituting an acyltransferase domain of the erythromycin polyketide synthase. J Antibiot,2003, 56(6): 543−551.
[42] Stassi DL, Kakavas SJ, Reynolds KA, et al.Ethyl-substituted erythromycin derivatives produced by directed metabolic engineering. Proc Natl Acad Sci USA, 1998, 95(13): 7305−7309.
[43] Donadio S, McAlpine JB, Sheldon PJ, et al. An erythromycin analog produced by reprogramming of polyketide synthesis. Proc Natl Acad Sci USA,1993, 90(15): 7119−7123.
[44] Reid R, Piagentini M, Rodriguez E, et al. A model of structure and catalysis for ketoreductase domains in modular polyketide synthases.Biochemistry, 2003, 42(1): 72−79.
[45] Bevitt DJ, Staunton J, Leadlay PF. Mutagenesis of the dehydratase active site in the erythromycin-producing polyketide synthase.Biochem Soc Trans, 1993, 21(1): 30S.
[46] Cundliffe E, Bate N, Butler A, et al. The tylosin-biosynthetic genes of Streptomyces fradiae. Antonie Van Leeuwenhoek, 2001,79(3/4): 229−234.
[47] Xue YQ, Zhao LS, Liu HW, et al. A gene cluster for macrolide antibiotic biosynthesis in Streptomyces venezuelae: architecture of metabolic diversity. Proc Natl Acad Sci USA,1998, 95(21): 12111−12116.
[48] Waldron C, Matsushima P, Rosteck PR, et al.Cloning and analysis of the spinosad biosynthetic gene cluster of Saccharopolyspora spinosa. Chem Biol, 2001, 8(5): 487−499.
[49] Shah S, Xue Q, Tang L, et al. Cloning,characterization and heterologous expression of a polyketide synthase and P-450 oxidase involved in the biosynthesis of the antibiotic oleandomycin. J Antibiot, 2000, 53(5): 502−508.
[50] Long PF, Wilkinson CJ, Bisang CP, et al.Engineering specificity of starter unit selection by the erythromycin-producing polyketide synthase.Mol Microbiol, 2002, 43(5): 1215−1225.
[51] MacNeil DJ, Occi JL, Gewain KM, et al. Complex organization of the Streptomyces avermitilis genes encoding the avermectin polyketide synthase.Gene, 1992, 115(1-2): 119−125.
[52] Marsden AFA, Wilkinson B, Cortés J, et al.Engineering broader specificity into an antibiotic-producing polyketide synthase. Science,1998, 279(5348): 199−202.
[53] Gokhale RS, Hunziker D, Cane DE, et al.Mechanism and specificity of the terminal thioesterase domain from the erythromycin polyketide synthase. Chem Biol, 1999, 6(2):117−125.
[54] Kao CM, Luo GL, Katz L, et al. Engineered biosynthesis of a triketide lactone from an incomplete modular polyketide synthase. J Am Chem Soc, 1994, 116(25): 11612−11613.
[55] Jacobsen JR, Cane DE, Khosla C. Spontaneous priming of a downstream module in 6-deoxyerythronolide B synthase leads to polyketide biosynthesis. Biochemistry, 1998,37(14): 4928−4934.
[56] Cane DE. Programming of erythromycin biosynthesis by a modular polyketide synthase. J Biol Chem, 2010, 285(36): 27517−27523.
[57] Siskos AP, Baerga-Ortiz A, Bali S, et al.Molecular basis of Celmer's rules: stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases. Chem Biol,2005, 12(10): 1145−1153.
[58] Kallberg Y, Oppermann U, Jörnvall H, et al.Short-chain dehydrogenases/reductases (SDRs)-coenzyme-based functional assignments in completed genomes. Eur J Biochem, 2002,269(18): 4409−4417.
[59] Kao CM, Luo GL, Katz L, et al. Manipulation of macrolide ring size by directed mutagenesis of a modular polyketide synthase. J Am Chem Soc,1995, 117(35): 9105−9106.
[60] Rowe CJ, Böhm IU, Thomas IP, et al. Engineering a polyketide with a longer chain by insertion of an extra module into the erythromycin-producing polyketide synthase. Chem Biol, 2001, 8(5):475−485.
[61] Gokhale RS, Tsuji SY, Cane DE, et al. Dissecting and exploiting intermodular communication in polyketide synthases. Science, 1999, 284(5413):482−485.
[62] Kapur S, Lowry B, Yuzawa S, et al.Reprogramming a module of the 6-deoxyerythronolide B synthase for iterative chain elongation. Proc Natl Acad Sci USA, 2012,109(11): 4110−4115.
[63] Bourne Y, Henrissat B. Glycoside hydrolases and glycosyltransferases: families and functional modules. Curr Opin Struc Biol, 2001, 11(5):593−600.
[64] Schlunzen F, Zarivach R, Harms J, et al. Structural basis for the interaction of antibiotics with the peptidyl transferase centre in eubacteria. Nature,2001, 413(6858): 814−821.
[65] Agouridas C, Denis A, Auger JM, et al. Synthesis and antibacterial activity of ketolides(6-O-methyl-3-oxoerythromycin derivatives): a new class of antibacterials highly potent against macrolide-resistant and -susceptible respiratory pathogens. J Med Chem, 1998, 41(21):4080−4100.
[66] Zhang CS, Fu Q, Albermann C, et al. The in vitro characterization of the erythronolide mycarosyltransferase EryBV and its utility in macrolide diversification. ChemBioChem, 2007,8(4): 385−390.
[67] Lee HY, Chung HS, Hang C, et al. Reconstitution and characterization of a new desosaminyl transferase, EryCIII, from the erythromycin biosynthetic pathway. J Am Chem Soc, 2004,126(32): 9924−9925.
[68] Yuan YQ, Chung HS, Leimkuhler C, et al. In vitro reconstitution of EryCIII activity for the preparation of unnatural macrolides. J Am Chem Soc, 2005, 127(41): 14128−14129.
[69] Schell U, Haydock SF, Kaja AL, et al. Engineered biosynthesis of hybrid macrolide polyketides containing D-angolosamine and D-mycaminose moieties. Org Biomol Chem, 2008, 6(18):3315−3327.
[70] Tang L, McDaniel R. Construction of desosamine containing polyketide libraries using a glycosyltransferase with broad substrate specificity. Chem Biol, 2001, 8(6): 547−555.
[71] Sun P, Zhao QF, Yu FT, et al. Spiroketal formation and modification in avermectin biosynthesis involves a dual activity of AveC. J Am Chem Soc,2013, 135(4): 1540−1548.
[72] Liao RJ, Liu W. Thiostrepton maturation involving a deesterification-amidation way to process the C-terminally methylated peptide backbone. J Am Chem Soc, 2011, 133(9): 2852−2855.
[73] Duan L, Wang SF, Liao RJ, et al. Insights into quinaldic acid formation in thiostrepton biosynthesis facilitating fluorinated thiopeptide generation. Chem Biol, 2012, 19(4): 443−448.
[74] Chen DD, Zhang Q, Zhang QL, et al.Improvement of FK506 production in Streptomyces tsukubaensis by genetic enhancement of the supply of unusual polyketide extender units via utilization of two distinct site-specific recombination systems. Appl Environ Microbiol, 2012, 78(15): 5093−5103.
[75] Zhao QF, Wang M, Xu DX, et al. Metabolic coupling of two small-molecular thiols programs the biosynthesis of lincomycin A. Nature, 2015,518(7537): 115−119.
猜你喜欢
保健与生活(2022年7期)2022-04-08
华声文萃(2021年11期)2021-11-15
天津中医药(2020年5期)2020-06-01
国际呼吸杂志(2019年4期)2019-03-12
临床医药文献杂志(电子版)(2017年12期)2017-05-18
中成药(2017年4期)2017-05-17
中国民族医药杂志(2016年6期)2016-05-09
浙江中西医结合杂志(2016年2期)2016-01-25
应用化工(2014年5期)2014-08-08
中国药物经济学(2013年4期)2013-06-07
