
苯胺及掺杂态苯胺低聚物结构的密度泛函理论研究
2015-03-23 11:56薛严冰于婧怡唐祯安
原子与分子物理学报 2015年2期
薛严冰, 于婧怡, 许 芝, 唐祯安
(1.大连交通大学电气信息学院, 大连 116028; 2.大连理工大学电子科学与技术学院, 大连 116023)
苯胺及掺杂态苯胺低聚物结构的密度泛函理论研究
薛严冰1, 于婧怡1, 许 芝1, 唐祯安2
(1.大连交通大学电气信息学院, 大连 116028; 2.大连理工大学电子科学与技术学院, 大连 116023)
采用基于密度泛函理论的计算方法,研究了苯胺低聚物及盐酸、对甲苯磺酸掺杂苯胺低聚物的几何结构和电子结构.结果表明,质子酸掺杂使苯胺低聚物分子链上醌环中C-C单、双键交替的性质被削弱,同时链间C=N键长明显增大.掺杂位上链间C-N-C键角增大,相邻环间的扭转角减小,分子链的共面性有所改善.与盐酸掺杂对比,对甲苯磺酸掺杂更利于电子从环内向链间转移,理论上可更好地改善聚苯胺材料的导电性能.
苯胺低聚物; 密度泛函法; 几何结构; 电子结构
1 引 言
聚苯胺(Polyaniline,PANI)是一种典型的导电高分子材料,具有原料易得、合成简单、良好的环境稳定性、导电性、光电性、热电性等诸多优点,近几年已引起研究者的广泛关注.有关其物理、化学性质、制备方法及应用等的实验研究已取得一定进展[1-4],但从理论计算角度出发,着眼于聚苯胺分子的微观结构及导电机理的研究还相对较少.
Lim S. L.等[5]是最早采用量子计算方法研究聚苯胺材料特性的.他们分别采用基于第一性原理(ab initio)和密度泛函理论(DFT)计算的方法研究了几种中性苯胺低聚体的几何和电子结构,研究结果表明用DFT方法得到的计算结果同由X射线衍射、紫外光吸收谱和X射线光电子能谱等实验观察到的数据很好地保持一致.此后陆续有采用第一性原理和(或)DFT计算方法研究苯胺低聚体的成果报道[6-16].Foreman J. P.等[7]用DFT的方法研究了樟脑磺酸(CSA)掺杂的还原结构苯胺低聚体,发现胺和磺酸基团间的氢键增加了苯-氮-苯骨架转移电子密度的能力.Yang G.等[14]分析了盐酸和樟脑磺酸掺杂的苯胺低聚体的电子结构,发现后一种掺杂较前一种掺杂能引起更多的电荷转移.Casanovas J.等[15]用DFT方法对聚苯胺低聚体进行研究,他们建立了从甲基到戊基不同的烷基硫酸盐阴离子,链上包含2-6个苯环的低聚体模型,系统研究了掺杂烷基的长度和聚合物链模型的大小对计算结果的影响.
由于计算模型、计算方法、计算参数等的不同,上述研究结果并不完全具有可比性.特别是针对掺杂态聚苯胺,已有的研究只涉及到盐酸和樟脑磺酸掺杂.有研究表明对甲苯磺酸(P-TSA)掺杂可提高聚苯胺材料对某些气体的响应灵敏度,有望实现其在气敏材料方面的应用[17],而有关P-TSA掺杂苯胺低聚体的理论计算尚未见有文献报道.本文采用基于密度泛函理论的计算方法,建立本征聚苯胺低聚体模型及盐酸和P-TSA掺杂聚苯胺低聚体模型,研究并比较不同酸掺杂态苯胺低聚体的几何结构和电子结构,提出质子酸掺杂改善聚苯胺导电性能的机理,为有针对性地设计导电聚苯胺材料提供理论依据.
2 模型与计算方法
2.1 理论模型
PANI目前广为接受的分子链模型是1986年MacDiarmid提出的苯式-醌式结构单元共存的模型[18]:
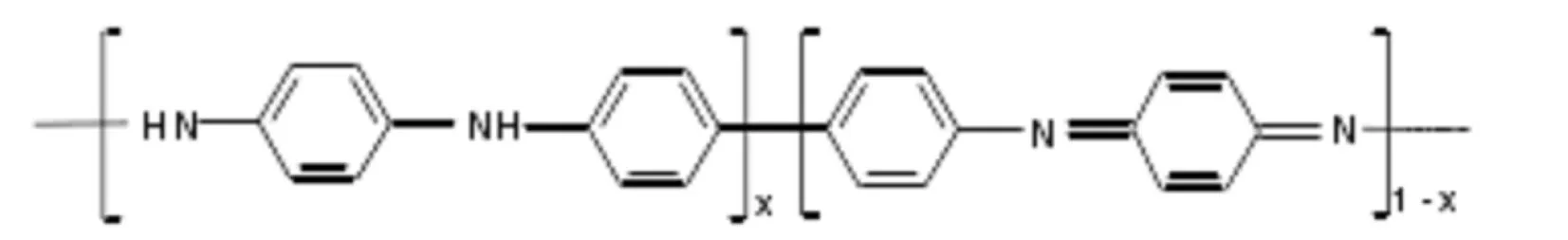
式中x代表PANI的氧化程度,当x=1时为完全还原态的全苯式结构,x=0时为完全氧化态的苯-醌交替结构;而x=0.5为苯-醌比为3:1的半氧化结构.在上述3种可稳定存在的PANI形态中,只有半氧化态PANI能通过掺杂发生从绝缘态到导电态的突变[19],因此本文以半氧化态PANI作为初始计算模型.
2.2 计算方法
采用基于密度泛函的理论计算方法,应用Accelrys 公司Material Studio软件包中的Dmol3计算模块进行计算[20].参数设置为:采用广义梯度近似(GGA),PBE泛函[14],结构优化采用BFGS计算方法,电子设置为全电子方式,基组选择为极化的双数值基组(DNP),SCF收敛判据设为中等精度的收敛条件.为加快收敛速度,选用了DIIS算法.先对建好的分子构象进行几何优化,再进行电子特性计算.
3 结果与讨论
3.1 PANI低聚物分子链结构的确定
分别建立了包含4、8、12、16个芳环的半氧化态苯胺低聚体模型,研究芳环数对分子链几何结构的影响.几何参数包括键长(环内的C-C键长及环间的C-N键长)、C-N-C键角及相邻两环之间的扭转角.计算结果表明,苯环内C—C单键键长在0.1395 nm-0.1454 nm之间,C=C双键键长在0.1382 nm-0.1415nm之间,单键键长缩短而双键键长拉长,说明形成了共轭长链体系[21].芳环数增加对C-C键长的影响不明显,不同链长度的C-C平均键长基本不变.芳环数增加对C-N键长有一定影响,C—N平均长度由4环的0.1384 nm增加至16环的0.1392 nm,C=N平均键长也由4环的0.1313 nm增加至16环的0.1323 nm.随环数增加,C-N-C键角平均值有增加趋势(图1(a)),相邻环间扭转角有减小趋势(图1(b)).当环数由12个增加到16个时,键角平均值趋于稳定,约为126°,扭转角平均值亦基本保持恒定,约为41.7°.
上述结果说明,当聚合物包含16个芳环时,体系的结构已趋于稳定,比较接近真实的聚合物片段.考虑到环数近一步增加时,会带来计算时间的成倍增加,故选择分子链模型为16环聚合物.
3.2 HCl掺杂态苯胺低聚物
图2是HCl掺杂苯胺低聚物的计算模型.首先建立HCl分子模型并几何优化,优化后H—Cl键长为0.1285 nm,比理论值键长0.127 nm略长;键能为425.2 KJ/mol,比理论键能431 KJ/mol 略小.按照优化后的分子结构,将HCl分子以H原子靠近醌环两侧N原子的方式置于苯胺低聚体链上.掺杂方式采用双极化子掺杂方式[9],即醌环两侧的N原子均为掺杂点.
3.2.1 几何结构
经优化得到苯胺低聚物的稳定构型.可以发现分子链具有规则的扭转性,同时H—Cl键断开,H质子与亚胺基的氮原子成键.HCl掺杂前后苯胺低聚物C-C键长的计算结果如图3(a)所示.掺杂对苯环中C-C键长影响不明显,却很大程度上改变了醌环中的C-C键长值.醌环中C-C单、双键交替的性质被明显削弱,醌环有被还原为苯环的趋势.图3(b)为HCl掺杂前后苯胺低聚物C—N键长的计算结果.同掺杂前相比,掺杂位置上C—N单键长基本保持不变,而C=N双键键长则明显增加,平均键长由掺杂前的0.1323 nm增加至0.1357 nm.说明H质子的加入对醌环上C-N键的影响远大于对苯环上的C-N键.
图4(a)是HCl掺杂前后聚合物的C-N-C键角的计算值.经分析,未掺杂点的C-N-C键角略微减小,平均值由129.37°减小至128.71°;而掺杂点位置上的C-N-C键角明显的增加,平均值由123.97°增加至129.39°,这一结论和文献[7,14]的计算结果保持一致.可能的原因是由于氨基N原子同掺入的H质子间形成氢键,N的孤对电子转移到掺杂物和聚苯胺骨架中,使C-N-C键角增加.
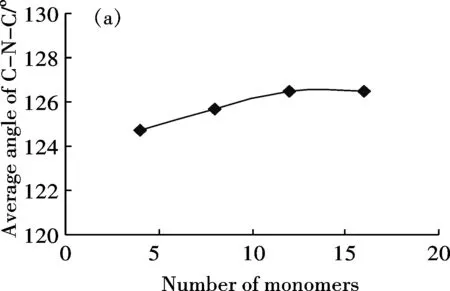
(a)Average angle of C-N-C bond
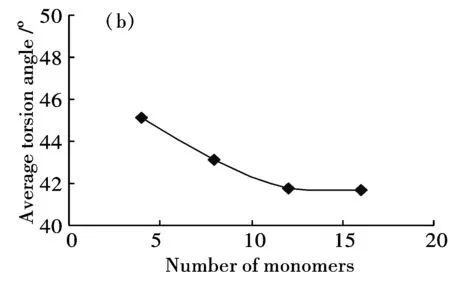
(b)Average torsion angle
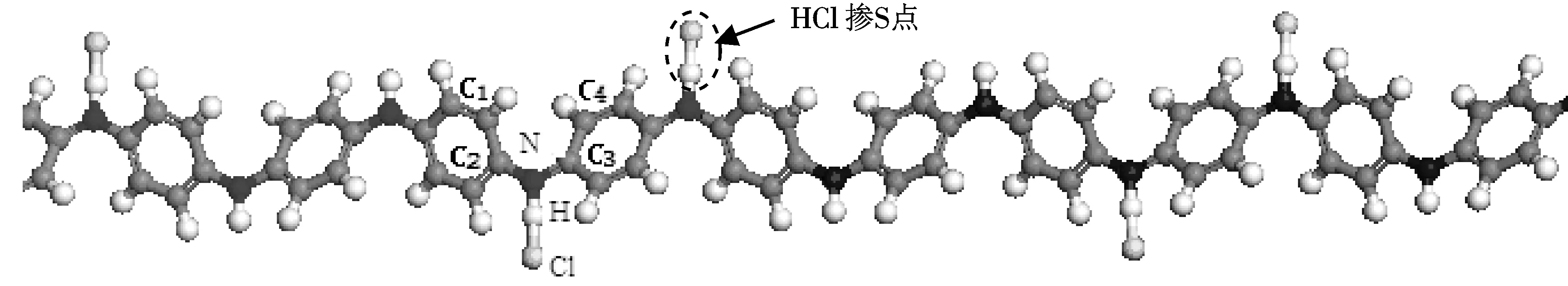
图 2 HCl掺杂苯胺低聚物的计算模型Fig.2 Calculation models of HCl doped Oligoaniline
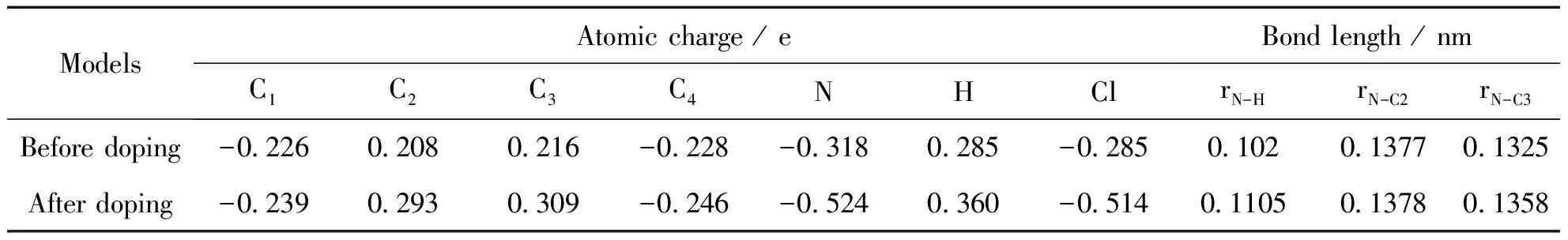
表1 HCl掺杂点附近的原子电荷和键长
HCl掺杂前后聚合物相邻芳环之间扭转角的计算结果图4(b).通常情况下,分子平面性越好,分子间的轨道重叠程度越高,其导电能力也越强.可以发现,掺杂位置上相邻芳环间的扭转角明显减小,平均扭转角由41.57°减小到33.94°.扭转角代表着N原子的Pz轨道同芳环中π电子的共轭程度,掺杂后相邻环的共面性有所改善,体系π共轭效果增加,电子易于沿着芳环的骨架进行离域.
3.2.3 电子结构
按照图2(a)中HCl掺杂点附近的原子标号,表1列出掺杂前后体系内各原子的原子电荷计算结果,表中的数值为8个掺杂点计算结果的平均值.
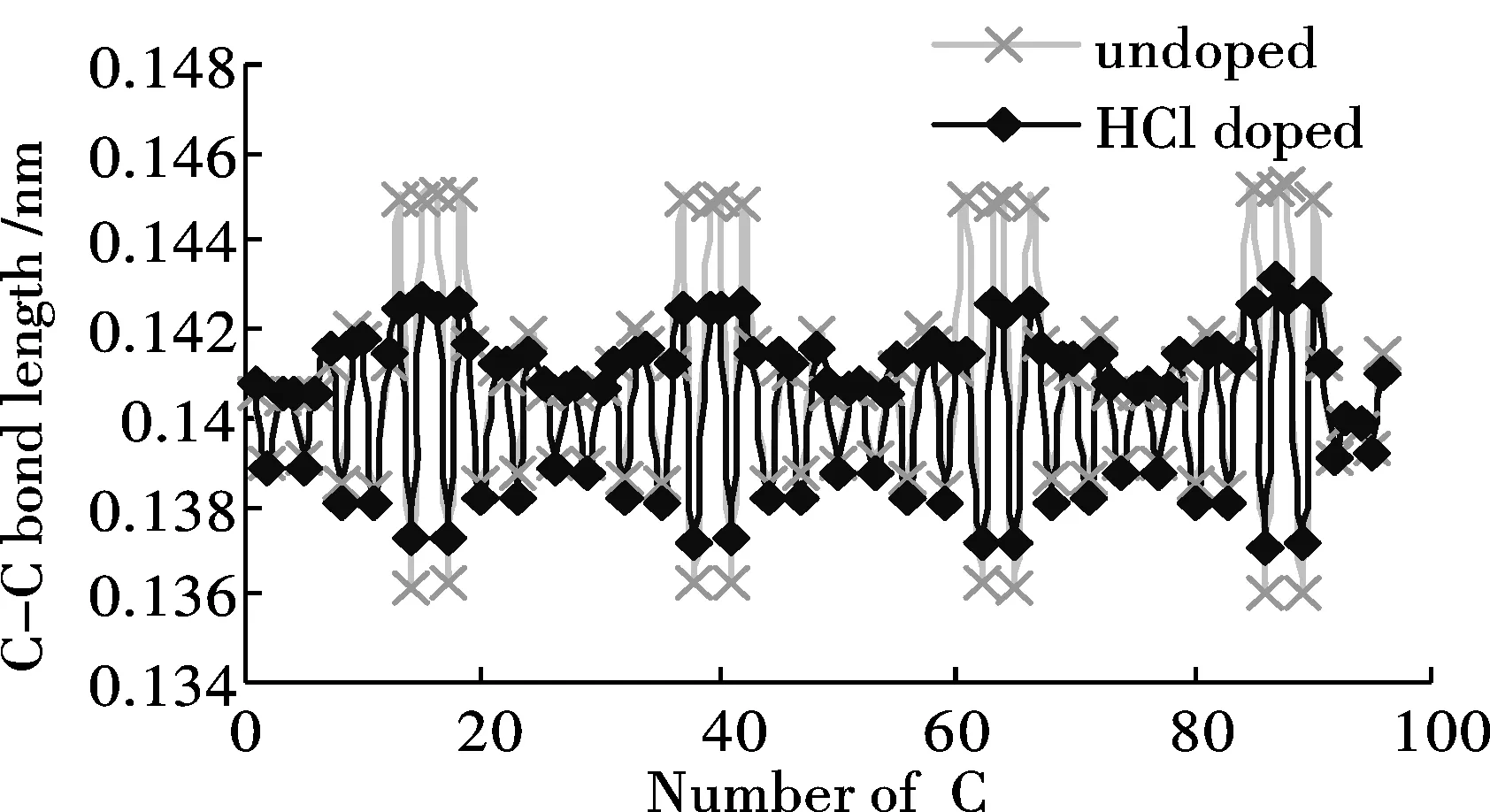
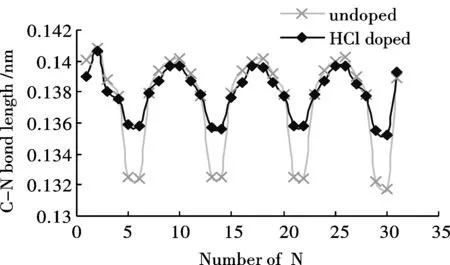
图 3 HCl掺杂对低聚物键长的影响Fig.3 Influence of HCl doping on the bond lengths of Oligoaniline


图4 HCl掺杂对低聚物键角的影响Fig.4 Influence of HCl doping on the bond angles of Oligoaniline
从表中可以看出,掺杂后C2和C3原子的缺电性增加,而N原子电子云密度增加,其原子电荷QN从-0.318 e变为-0.524 e,说明掺杂使电子从芳环转移到链上N原子.由于在掺杂位形成氢键,存在电子在掺杂物和底物之间的转移,造成聚苯胺环内电子的重新分布,电子密度的转移是产生几何构型变化的根本原因.原子电荷的计算结果很好的验证了几何计算结果,由于电子从环内转向链间,使链间C-N-C的键角增加,相邻环之间的两面角减小,电子的共轭相应增强,从而提高了聚苯胺的导电性能.从N—H键长的计算结果可以看出,掺杂点形成的氢键长度为0.1105 nm,大于苯环中的氢键长度0.102 nm,说明掺杂形成的氢键强度较弱,易于通过去质子化实现去掺杂.
3.3 对甲苯磺酸掺杂态聚苯胺
按照P-TSA结构式绘制了P-TSA分子,在对分子进行结构优化的基础上,将P-TSA分子中磺酸基中的H原子置于苯胺醌环两侧的N原子上,构建了P-TSA掺杂PANI模型,如图5所示.为了减小掺杂大分子对苯胺结构产生空间位阻效应,建模时尽量使P-TSA分子中的苯环远离聚苯胺分子链.为了同HCl掺杂作对比,掺杂方式同样采用双极化子掺杂.
3.3.1 几何结构
P-TSA掺杂对聚苯胺中各键键长影响的规律同HCl掺杂一致,表现为醌环中的C=C双键键长明显增加,同时连接各环的C=N双键键长亦明显增加,说明掺杂后聚合物链原本具有的苯-醌交替的结构不复存在,更多地体现出一种芳香环的共轭结构.
由于掺杂对非掺杂点芳环几何结构的影响很小,表2仅列出了各掺杂位置上C-N-C键角和相邻环扭转角的平均值计算结果.掺杂使C-N-C键角增大,同时使相邻环间的扭转角减小.对比具体的数值,可发现P-TSA掺杂方式下C-N-C键角的增加略大于HCl掺杂,共面性也略好于盐酸,因此推测P-TSA掺杂比HCl掺杂具有更好的导电特性.
3.3.2 电子结构
P-TSA掺杂前后体系内各原子的原子电荷计算结果如表3所示,表中的原子标号见图5.表中列出的数据为8个掺杂点计算结果的平均值.
P-TSA分子掺杂后,原分子中磺酸根与H原子形成的氢键断裂,H原子同苯胺低聚物中的亚胺基氮结合形成氢键.从与P-TSA分子相关的原子电荷变化可以看出,磺酸中的H原子与亚胺基N原子形成氢键后,H的原子电荷从0.435e增加至0.458 e,说明掺杂后形成的氢键中,H的正离子型更强.磺酸分子中S原子电荷从0.637e减小至0.625 e,而与其成键的三个O原子电荷均有所增加,说明失去氢质子后磺酸根中S—O键的电子密度向O原子一边偏移.重点考察掺杂点上的N原子,发现N的原子电荷由-0.318 e变化至-0.593 e,即掺杂使N原子的电子云密度增加0.275 e,而与氮相邻的C2、C3原子则失去电子,说明N原子得到的电子是由苯环中的π电子提供的.与HCl掺杂后N原子电荷对比(变化量为0.206 e),可看出有机磺酸掺杂更利于电子从环内向链间转移.原子电荷的计算结果同几何优化结果一致,均从理论上说明PANI采用P-TSA掺杂可比HCl掺杂获得更好的导电特性.
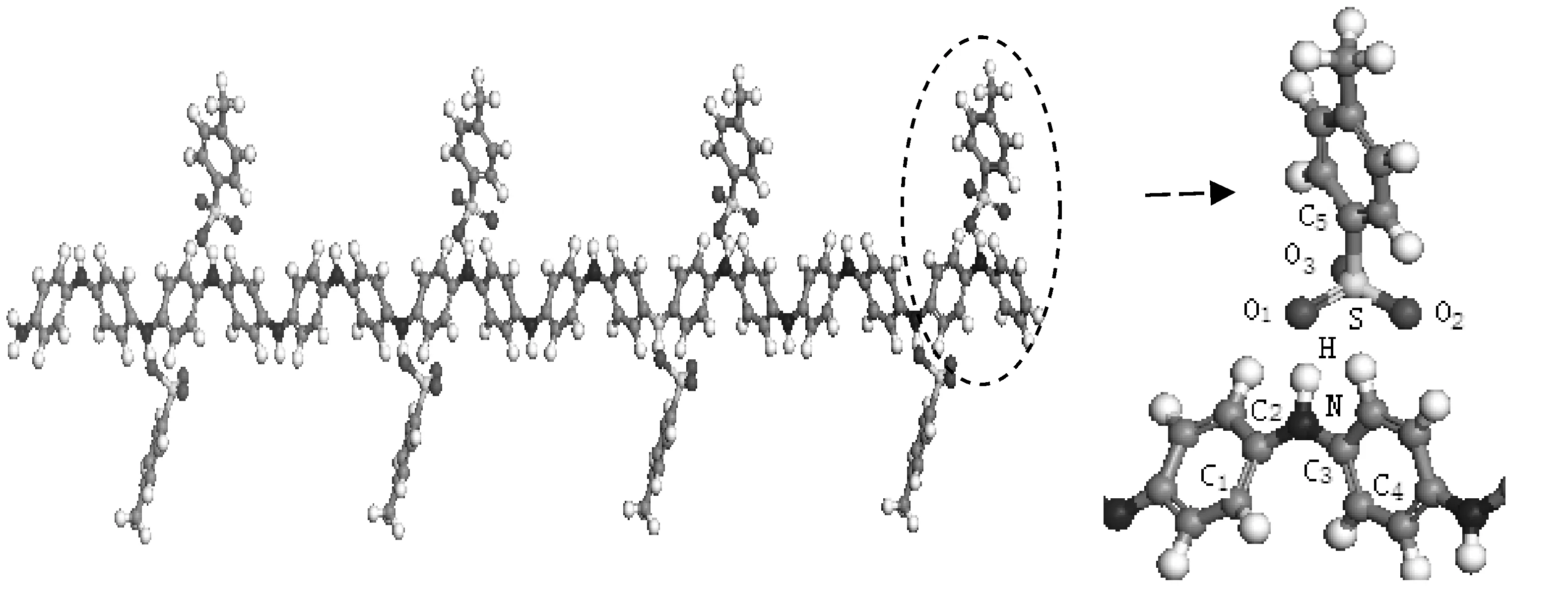
图5 P-TSA掺杂苯胺低聚物模型Fig.5 Model of P-TSA doped Oligoaniline
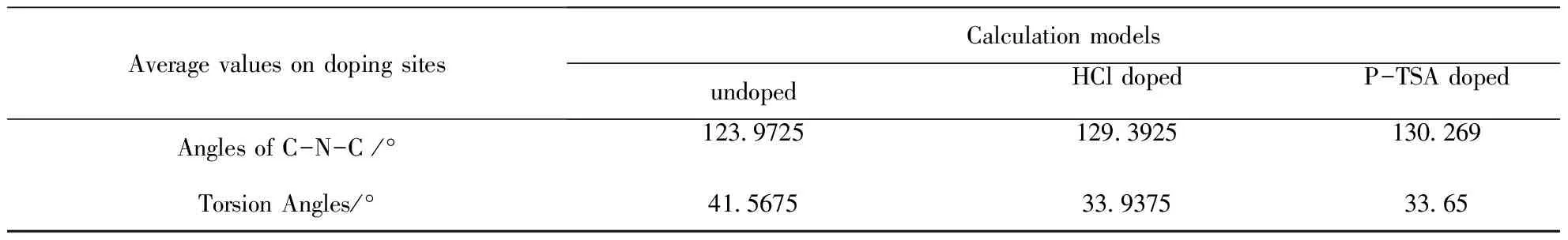
表2 P-TSA掺杂位上C-N-C键角和扭转角的计算结果
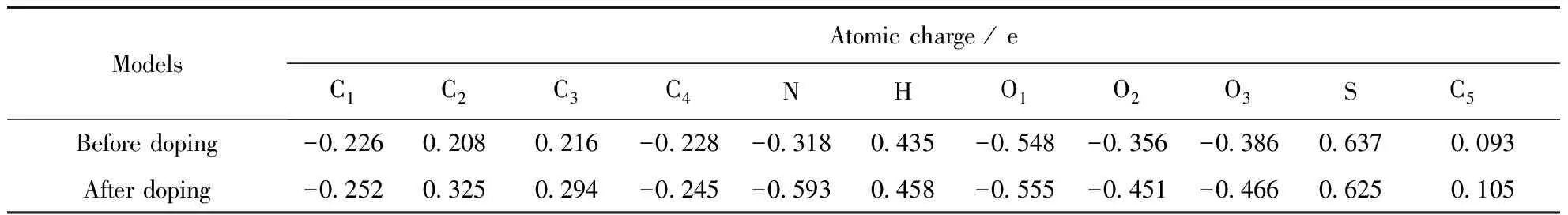
表3 P-TSA掺杂点附近的原子电荷
4 结 论
采用密度泛函理论方法,对苯胺低聚物及质子酸掺杂苯胺低聚物的结构进行了计算与分析.结果表明:(1)HCl掺杂很大程度上改变了醌环中的C-C键长,C-C单、双键交替的性质被削弱.掺杂位置上C=N双键平均键长由0.1323 nm增加至0.1357 nm,链间C-N-C键角平均值由123.97°增至129.39°,扭转角平均值由41.57°减小到33.94°.掺杂位N原子电荷减少,而与其成键的两个C原子电荷均有所增加,电子从芳环转移到链上.(2)P-TSA掺杂改变苯胺低聚物几何结构的规律同HCl掺杂一致,掺杂点C-N-C键角平均值略大于HCl掺杂,相邻环间的共面特性略优于HCl掺杂.P-TSA掺杂点上N原子电荷变化0.275 e,大于HCl掺杂的0.206 e,理论上表明有机磺酸掺杂更利于电子从环内向链间转移.
[1] Rajesh Tarushee A, Devendra K. Recent progress in the development of nano-structured conducting polymers /nanocomposites for sensor applications [J].SensorsandActuatorsB, 2009, 136: 275.[2] Masanobu M, Takuya A. Properties and stability of polyaniline nanofiber ammonia sensors fabricated by novel on-substrate method [J].SensorsandActuatorsB, 2011, 160: 999.
[3] Tai H L, Jiang Y D, Xie G Z,etal. Preparation, characterization and comparative NH3-sensing characteristic studies of PANI/inorganic Oxides nanocomposite thin films[J].JournalofMaterialsSciencesandTechnology, 2010, 7: 605.
[4] Liu H Y, Wang S B, Zhu Y,etal. Hydrothermal synthesis of polyaniline-iron oxide nano-composite materials [J].ChemicalJournalofChineseUniversities, 2013, 34(5): 1303 (in Chinese) [刘红缨, 王双宝, 朱英, 等. 水热法制备聚苯胺及其铁氧化物纳米复合材料[J]. 高等学校化学学报, 2013, 34(5): 1303]
[5] Lim S L, Tana K L. A comparative ab initio and DFT study of neutral aniline oligomers [J].JournalofChemicalPhysics, 2000, 122(23): 10648.
[6] Cavazzoni C, Colle R, Farchioni R,etal. Car-Parrinello molecular dynamics study of electronic and structural properties of neutral polyanilines [J].PhysicalReviewB, 2002, 66: 165110.
[7] Foreman J P, Monkman A P. Theoretical investigations into the structural and electronic influences on the hydrogen bonding in doped polyaniline [J].TheJournalofPhysicalChemistryA, 2003, 107: 7604.
[8] Varela A, lvarez A, Sordo J,etal. Doping of polyaniline by acid-base chemistry: Density functional calculations with periodic boundary conditions [J].TheJournaloftheAmericanChemicalSociety, 2005, 127:11318.
[9] Casanovas J, Aleman C. Comparative theoretical study of heterocyclic conducting oligomers neutral and oxidized forms [J].TheJournalofPhysicalChemistryC, 2007, 111: 4823.
[10] Zhekova H, Tadjer A, Ivanova A,etal. Theoretical study of the structure and electronic spectra of fully protonated emeraldine Oligomers [J].InternationalJournalofQuantumChemistry, 2007, 107: 1688.
[11] Colle R, Parruccini P, Benassi A,etal. Optical properties of emeraldine salt polymers from ab initio calculations: Comparison with recent experimental data. [J].TheJournalofPhysicalChemistryB, 2007, 111: 2800.
[12] Cavazzoni C, Colle R, Farchioni R,etal. Acidification of three-dimensional emeraldine polymers: Search for minimum energy paths from base to salt [J].JournalofChemicalPhysics, 2008, 128: 234903.
[13] Aleman C, Ferreira C A, Torras J,etal. On the molecular properties of polyaniline: A comprehensive theoretical study [J].Polymer, 2008, 49: 5169.
[14] Yang G, Hou W, Feng X O,etal. Electronic structure of oligoaniline doped by inorganic and organic acids [J].InternationalJournalofQuantumChemistry, 2008, 108: 1155.
[15] Casanovas J, Canales M, Ferreira C A,etal. A first principle analysis of the structure of oligoanilines doped with alkylsulfonic acids [J].TheJournalofPhysicalChemistryA, 2009, 113: 8795.
[16] Zhang H F, Zhang S D, Kong X H. Vibrational spectrum of C6H5NH2in S1state and ab initio calculations[J].JournalofAtomicandMolecularPhysics(原子与分子物理学报), 2010, 27(4): 643.
[17] Paterno L G, Mattoso L H C. Influence of different dopants on the ddsorption, morphology, and properties of self-assembled films of Poly(o-ethoxyaniline) [J].JournalofAppliedPolymerScience, 2002, 83: 1309.
[18] MacDiarmid A G, Chiang J C, Richter A F,etal. Polyanilne: a new concept in conducting polymers [J].SyntheticMetals, 1987, 18(1-3): 285.
[19] MacDiarmid A G, Epstein A J. Secondary doping in polyanilne[J].SyntheticMetals, 1995, 69: 22.
[20] Accelrys Incorporation, Material Studio, Accelrys Inc: SanDiego, CA.
[21] Zhang F L, Wu X F. Density functional theory study of 3-octylthien-2, 5-ylenediethynylene-co- benzo[c]- 1’2’ 5’- thiadiazo-3, 6-ylenedi adsorption on TiO2(100) Surface[J].JournalofAtomicandMolecularPhysics, 2012, 29(5): 927 (in Chinese) [张福兰, 吴兴发. 4, 7-二(2-噻吩基)苯并噻二唑-3-辛基噻吩二炔在TiO2(100)表面吸附的密度泛函理论研究[J].原子与分子物理学报, 2012, 29(5): 927]
Density functional theory study of the structures of oligoaniline and doped-oligoaniline
XUE Yan-Bing1, YU Jing-Yi1, XU Zhi1, TANG Zhen-An2
(1. School of Electrical and Information, Dalian JiaoTong University, Dalian 116028, China 2.School of Electronic Science and Technology, Dalian University of Technology, Dalian 116024, China)
A comprehensive study about the geometric structure and electronic structure of aniline oligomer and hydrochloric acid, p-toluene sulfonic acid doped oligomer has been developed using theoretical calculation method based on density functional theory. The results show that the property of C-C bond length of quinone ring in Oligoaniline molecular chains alternating in the way of single and double bond is obviously weakened by doped proton-acid, and the bond length of C=N between chains is significantly increased. The bond angle of C-N-C on doping site is increased and the torsion angle between adjacent rings is reduced. Coplanar of the molecular is improved by acid doping. Compared with HCl, doping with p-toluene sulfonic acid is more conducive to transfer electron from the aromatic rings to the molecular chains, so doping with organic acid can better improve the conductivity of Polyaniline material theoretically.
Oligoaniline; Density functional method; Geometrical structure; Electronic structure
103969/j.issn.1000-0364.2015.02.005
2013-11-25
国家自然科学基金(61201092);辽宁省自然科学基金(201202015);辽宁省高等学校优秀人才支持计划(LJQ2013047)
薛严冰,女,博士,教授,硕士生导师,主要从事半导体气体传感器方面的研究. E-mail: dlxyb@djtu.edu.cn
O649.5
A
1000-0364(2015)02-0201-06
猜你喜欢
能源化工(2022年1期)2023-01-14
能源化工(2021年6期)2021-12-30
纺织检测与标准(2021年3期)2021-12-03
昭通学院学报(2021年5期)2021-03-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
中国塑料(2015年7期)2015-10-14
中国塑料(2014年11期)2014-10-17
中国塑料(2014年8期)2014-10-17
