
尿蛋白及晚期糖基化终产物对肾小管上皮细胞溶酶体的影响*
2015-04-17 07:53邓健锟王淑君吴洪銮罗勉娜许碧华潘庆军刘华锋刘伟敬
中国病理生理杂志 2015年3期
邓健锟, 王淑君, 吴洪銮, 罗勉娜, 许碧华, 梁 东, 潘庆军, 刘华锋, 刘伟敬
(广东医学院附属医院肾脏疾病研究所,广东 湛江 524001)
尿蛋白及晚期糖基化终产物对肾小管上皮细胞溶酶体的影响*
邓健锟▲, 王淑君▲, 吴洪銮, 罗勉娜, 许碧华, 梁 东, 潘庆军, 刘华锋, 刘伟敬△
(广东医学院附属医院肾脏疾病研究所,广东 湛江 524001)
目的: 探讨慢性肾脏病(CKD)病程中所产生的病理产物尿蛋白及晚期糖基化终产物(AGE)对肾小管上皮细胞(tubular epithelial cells,TECs)溶酶体结构与功能的影响,为阻止或延缓CKD病情进展探索新思路。方法: 临床上选取未经特殊治疗的微小病变肾病综合征(MCNS)患者(n=11)、糖尿病肾病(DN)患者(n=11)及活检基本正常(n=6)的肾组织标本,以溶酶体相关膜蛋白1(lysosomal-associated membrane protein 1,LAMP1)和组织蛋白酶B(cathepsin B,CB)行间接免疫荧光染色;体外以8 g/LJ尿蛋白或100 mg/L晚期糖基化终产物刺激人肾小管上皮细胞系(HK-2细胞),以LAMP1和CB行间接免疫荧光染色,检测 CB及组织蛋白酶L (cathepsin L,CL)活性,并观察DQ-卵清蛋白降解情况。结果: MCNS及DN患者TECs存在溶酶体膜透化(lysosomal membrane permeabilization,LMP)现象。与正常对照组相比,尿蛋白及AGE-BSA均可致HK-2细胞LMP发生率上升,CB及 CL活性降低,溶酶体对DQ-卵清蛋白的降解能力降低(P<0.05)。结论: CKD病理产物尿蛋白和晚期糖基化终产物均可致TECs出现LMP现象,并使溶酶体消化功能下降,这可能是CKD肾小管间质纤维化进展的重要机制之一。
肾小管上皮细胞; 溶酶体; 尿蛋白; 晚期糖基化终产物
现已基本明确,肾小球疾病患者肾小管-间质的损害程度与肾功能毁损程度高度正相关,其危害甚于肾小球损伤[1]。对于肾小球疾病患者,除了原发性肾小球损伤,继发性肾小管上皮细胞(tubular epithelial cells,TECs)损伤处于肾小管-间质纤维化整个病理生理过程的上游环节,对整个慢性肾衰竭病程的进展具有“承上启下”的作用[2]。因此,控制TECs损伤是阻止或延缓肾小管-间质纤维化进展的关键策略之一,而且对控制慢性肾衰竭整个病程的进展也有极其重要的意义。引起TECs损伤的原因甚多,对于我国慢性肾衰竭最主要病因的原发性肾小球疾病而言,尿蛋白作为肾小球损害的病理产物,是继发性肾小管损害的首要原因[3]。随着社会发展和人们生活方式改变,近年来糖尿病肾病(diabetic nephropathy,DN)也已成为慢性肾衰竭又一主要病因,其病理产物糖基化终产物也是继发性肾小管损害的重要因素[4]。因此,尿蛋白和糖基化终产物是当前主要慢性肾脏病(chronic kidney disease,CKD)发病过程中的主要病理产物。
溶酶体为膜封闭细胞器,存在于胞质,含各种水解酶以完成对细胞成分和大分子的降解[5],细胞内许多降解途径如异噬、自噬等[6]都是通过溶酶体来执行。有研究报道溶酶体可降解重吸收的尿蛋白[7-8],溶酶体组织蛋白酶促进晚期糖基化终产物(advanced glycosylation end products,AGE)相关修饰蛋白的降解,减轻其细胞毒性[9-10],由此可见溶酶体-蛋白降解系统在一定程度上对TECs维持细胞稳态,对抗损伤因素,促进细胞存活具有重要意义。然而,CKD病程中产生的尿蛋白和糖基化终产物等病理产生对TECs溶酶体影响如何,目前未见研究报道。
材 料 和 方 法
1 材料
人肾小管上皮细胞系HK-2细胞(ATCC细胞库);胎牛血清及DMEM/F12细胞培养液(Gibco);AGE(Biovision);兔抗人溶酶体相关膜蛋白1(lysosmal-associated membrane protein 1 ,LAMP1)及鼠抗人组织蛋白酶B(cathepsin B, CB) I抗(Abcam);驴抗兔Alexa Fluor® 488及羊抗鼠Alexa Fluor® 594 II抗、DQ-卵清蛋白(Invitrogen);CB及组织蛋白酶L(cathepsin L, CL)试剂盒(Biovision);激光共聚焦显微镜(Leica)。
2 实验方法
2.1 肾组织标本采集 微小病变肾病综合征(minimal change nephrotic syndrome,MCNS)肾组织标本(n=11)来源于未经治疗且病程小于1个月、肾脏病理明确诊断为MCNS的患者;DN肾组织标本(n=11)来源于肾脏病理明确诊断为DN的患者;对照肾组织标本(n=6)来源于临床上以血尿为主要表现而肾组织活检为基本正常的患者。
2.2 尿蛋白提取 进入本研究的MCNS患者均经病理学确诊,无感染及并发症,且未使用肾上腺皮质激素和免疫抑制剂治疗,无可疑肾毒药物(包括中药)应用史。收集MCNS患者的尿液,之后以硫酸铵沉淀法提取尿蛋白[11]。
2.3 细胞的培养及刺激 HK-2细胞以含10% 胎牛血清的DMEM/F12培养液,在37 ℃、5% CO2的条件下进行培养传代用于实验。经预实验调整药物刺激浓度和时间,确定以8 g/L尿蛋白刺激细胞16 h或100 mg/L AGE刺激细胞6 h。
2.4 免疫荧光法检测肾组织及 HK-2细胞LAMP1和CB的表达[12-13]收集病人肾组织标本制成冰冻切片,另胰酶消化细胞后制成细胞悬液,按2×108/L的密度将细胞接种于内含盖玻片的12孔板,待贴壁完全加入上述刺激物,刺激完毕收集各组HK-2细胞爬片。4%多聚甲醛固定,0.25% PBS-Triton X-100中通透,5% BSA封闭抗原后,滴加用2.5% BSA稀释的兔抗人LAMP1的 I抗(1∶200)和鼠抗人CB的I抗(1∶600), 4 ℃孵育过夜。次日PBS浸洗后,滴加驴抗兔Alexa Flour® 488的 II抗(1∶300)和羊抗鼠594 的II抗(1∶300)37 ℃孵育60 min,避光操作,PBS浸洗后滴加DAPI染核5 min后再次浸洗,封片后通过激光共聚焦显微镜检测。每次实验至少统计200个HK-2细胞,计算CB弥散细胞数量占总细胞数量百分比。
2.5 生物化学法检测酶的活性 以CB和CL试剂盒检测细胞培养上清中的CB和CL的水平,具体操作步骤严格按照试剂盒说明书。
2.6 溶酶体降解能力检测 细胞经尿蛋白及AGE刺激后,在加有10 mg/L DQ-卵清蛋白的培养液中共培养2 h,经PBS洗涤及4%多聚甲醛固定,之后以激光共聚焦显微镜检测。每次实验至少统计30个细胞,统计每一个HK-2胞浆的DQ点数量(绿色荧光点),计算实验组与对照组倍数比。
3 统计学处理
应用SPSS 16.0统计软件进行统计分析。实验数据用平均值±标准差(mean±SD)表示。两样本比较采用独立样本t检验;多个样本均数的比较采用单因素方差分析(one-way ANOVA);多个样本均数间每两个均数的比较首先用方差齐性检验,当总体方差齐时,选择Bonferroni法;当方差不齐时,选择Tamhane’s T2法。以P<0.05表示差异有统计学意义。所有实验至少重复3次。
结 果
1 尿蛋白及AGE致溶酶体膜透化发生
对照肾组织肾小管上皮细胞CB和LAMP1呈点状分布,且多数共定位存在;而MCNS患者肾组织及糖尿病肾病患者肾组织多数TECs 内CB弥散至整个胞浆,LAMP1排列不规则,见图1。体外实验采用HK-2细胞进一步证实了以上结果。统计胞浆弥散分布CB荧光的细胞数目占细胞总数的百分比,与对照组相比,尿蛋白组及AGE-BSA组该比例上升,差异有统计学意义(P<0.01),见图2。
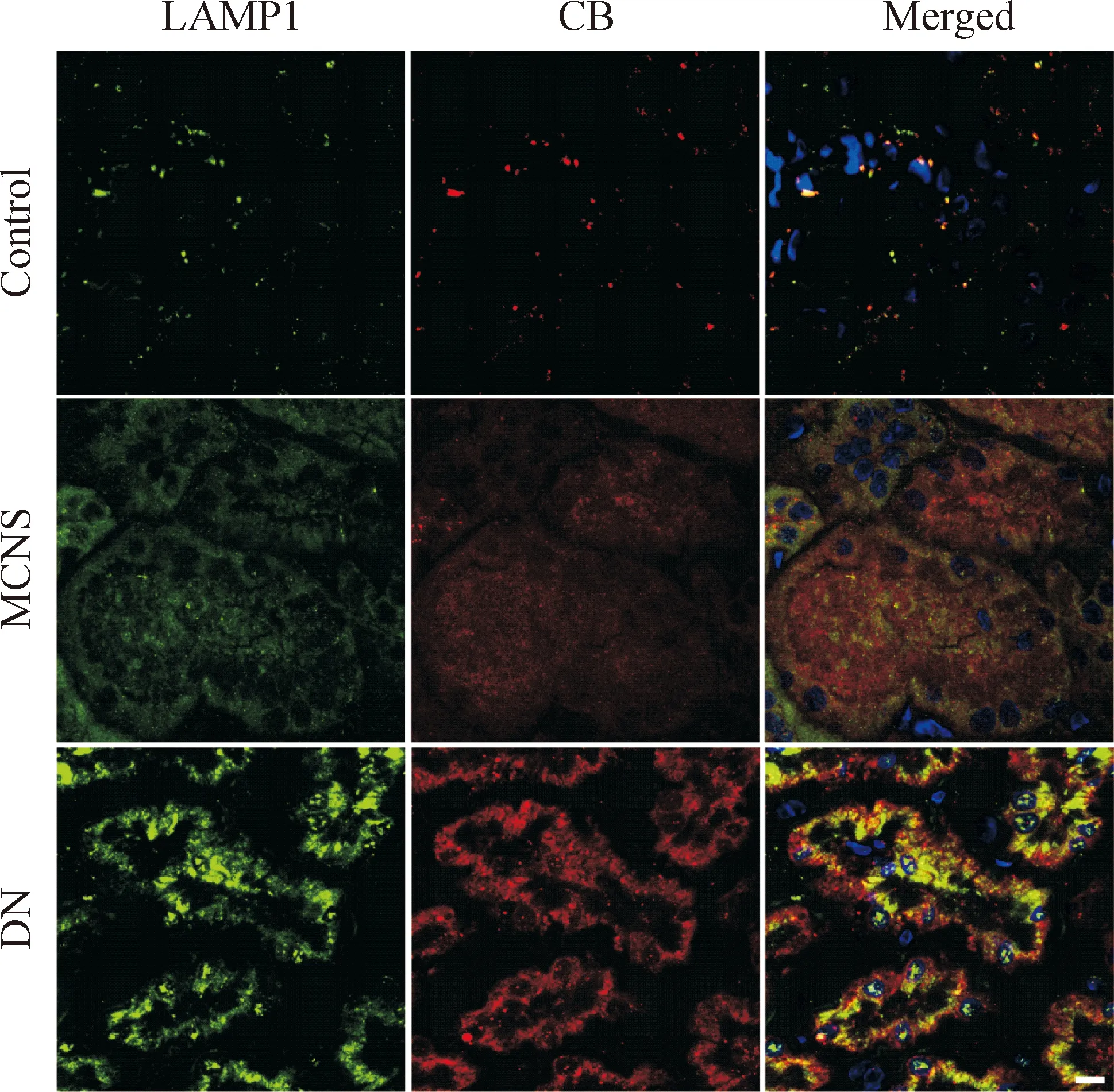
Figure 1.The distribution of CB and LAMP1 in TECs during the development of MCNS or DN. The scale bar=10 μm.
2 尿蛋白及AGE致溶酶体酶活性下降
如图3所示,尿蛋白及AGE刺激后的HK-2细胞溶酶体酶CB和CL活性均下降,与对照组相比差异有统计学意义。
3 尿蛋白及AGE致溶酶体对DQ-卵清蛋白降解能力下降
我们通过检测DQ-卵清蛋白荧光进一步明确溶酶体消化功能。如图4所示,与对照组相比,尿蛋白及AGE刺激后HK-2细胞DQ-卵清蛋白(绿色荧光)点数显著减少,差异有统计学意义(P<0.01)。
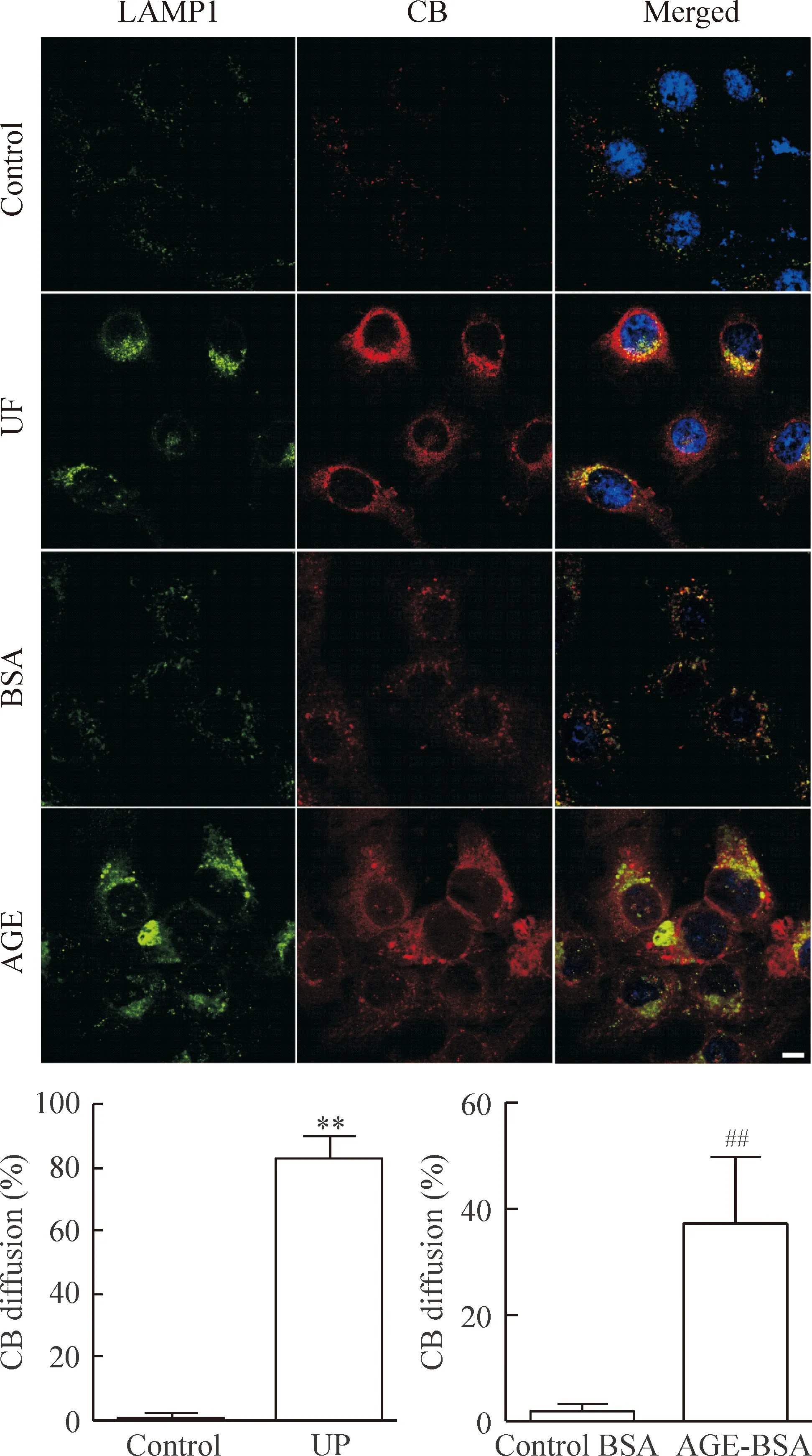
Figure 2.The distribution of CB and LAMP1 in the HK-2 cells after exposure to urinary proteins (UP, 8 g/L) for 16 h or AGE-BSA (100 mg/L) for 6 h . The scale bar=10 μm. Mean±SD. n=3.**P<0.01 vs control; ##P<0.01 vs control BSA.
讨 论
在本研究中,我们通过对某些酶和溶酶体膜进行间接免疫荧光双标记的方法,首次直接证实了CKD病程中所产生的病理产物尿蛋白和AGE可致溶酶体膜通透化(lysosomal membrane permeabilization,LMP)。LMP发生后,溶酶体内容物会透过溶酶体膜释放到细胞浆,这些释放至胞浆的溶酶体水解酶可非特异性消化细胞器;其次LMP作为细胞死亡信号传导重要组成部分已达成广泛共识,一旦溶酶体膜受损,泄露入胞浆的溶酶体酶可引起TECs发生细胞凋亡或自噬性死亡,而大量溶酶体破裂则可引起细胞坏死[12, 14-15]。溶酶体最重要的功能便是消化功能,溶酶体内各种组织蛋白酶是其发挥消化功能的主体[16]。既然溶酶体有LMP发生,其消化功能可能受到影响。我们首先对溶酶体内几种主要的蛋白水解酶的活性进行了分析,发现尿蛋白和AGE-BSA均可致小管上皮细胞溶酶体CB和CL活性均降低。我们进一步通过检测溶酶体对DQ-卵清蛋白的降解能力来说明其消化功能的变化。DQ-卵清蛋白是一种连接自我淬灭染料的卵清蛋白,此蛋白未被处理时不发出荧光,而当它被细胞摄入消化后会呈现强烈的绿色荧光。如结果所示,尿蛋白和AGE-BSA刺激下培养肾小管上皮细胞DQ-卵清蛋白荧光明显减少,即细胞降解DQ-卵清蛋白的能力下降,由此我们推断上述2种病理产物刺激下溶酶体消化功能是下降的。溶酶体作为溶酶体-蛋白降解系统的重要组成部分,其消化功能降低可能导致异噬-溶酶体途径和自噬-溶酶体途径的阻塞。当异噬-溶酶体途径堵塞时,贮存胞内降解受阻的尿蛋白可通过Bad[17]、内质网应激[18]、氧化应激[19-20]等多种途径引起细胞凋亡。而此时自噬-溶酶体途径的堵塞使得自噬无法发挥对抗尿蛋白所致TECs损伤的作用[21]。大量蛋白尿可通过削弱溶酶体消化功能导致TECs处理外来蛋白及降解自身受损蛋白的能力下降,进一步加重细胞损伤。早在2001年,有研究提示糖尿病致病因素可致肾脏溶酶体消化功能下降,其具体机制未明确,而溶酶体对白蛋白降解能力减弱与白蛋白尿的形成有着切密关系[22-23]。近几年,亦有报道溶酶体消化功能下降伴随着自噬流下降与DN发病机理相关[24]。另Shechter等[25]证明糖尿病条件下溶酶体损伤可致肾脏内多种蛋白降解受阻,导致基质堆积进而促进糖尿病肾脏肥大的进展,提示DN状态下溶酶体的消化功能受损与疾病进展密切相关。

Figure 3.The effects of urinary proteins (UP) or AGE-BSA on enzymatic activity of CB and CL in the HK-2 cells. Mean±SD. n=3. **P<0.01 vs control; #P<0.05, ##P<0.01 vs control BSA.

Figure 4.The effects of urinary proteins (UP) or AGE-BSA on the degradation of DQ-ovalbumin in the HK-2 cells. Mean±SD. n=3. ** P<0.01 vs control; ##P<0.01 vs control BSA.
持续病理产物(如尿蛋白或AGE)作用下,肾小管上皮细胞损伤(凋亡和活化)。一方面,TECs过度凋亡部分导致了肾小管萎缩;另一方面, TECs活化后分泌各种生长因子激活了肾间质的成纤维细胞而导致肾间质纤维化,活化的 TECs还可转分化为肌成纤维细胞,肌成纤维细胞又可分泌大量的细胞外胶原基质加剧肾间质纤维化,此外,TECs转分化后穿破肾小管基底膜游走进入肾间质,使TECs丢失而进一步加剧肾小管萎缩,最后,活化的TECs还可分泌各种趋化因子促使炎症细胞向间质浸润。以上这些病理生理变化可能参与CKD患者肾小管萎缩和间质纤维化[2]。
本研究观察了当前2种最常见CKD疾病过程中所产的2种最经典的病理产物对肾小管上皮细胞溶酶体结构和功能的影响,但仍然不足以全面说明CKD状态下TECs溶酶体的变化情况,因为CKD状态下还有许多病理产物,如白细胞介素[26],或尿毒症毒素[27]也都已被证实能对肾小管上皮细胞造成损害,它们是否也以相同方式造成TECs溶酶体损伤还有待进一步研究,而且它们之间作用是否叠加也值得进一步探讨。
本研究结果证实了CKD状态下病理产物尿蛋白及糖基化终产物等可导致肾小管上皮细胞溶酶体发生LMP等结构性损伤和消化功能降低等功能性损伤,较充分地证实了CKD状态下所产生的病理产物可能是肾小管-间质进一步损伤的下游病因,阻止这些病理产物对溶酶体的损伤用以改善溶酶体功能将可能是延缓CKD进展的新思路。
[1] Becker GJ, Hewitson TD. The role of tubulointerstitial injury in chronic renal failure [J]. Curr Opin Nephrol Hypertens, 2000, 9(2): 133-138.
[2] Nangaku M. Mechanisms of tubulointerstitial injury in the kidney: final common pathways to end-stage renal failure [J]. Intern Med, 2004, 43(1): 9-17.
[3] Zoja C, Benigni A, Remuzzi G. Cellular responses to protein overload: key event in renal disease progression[J]. Curr Opin Nephrol Hypertens, 2004, 13(1): 31-37.
[4] Morcos M, Sayed AA, Bierhaus A, et al. Activation of tubular epithelial cells in diabetic nephropathy[J]. Diabetes, 2002, 51(12): 3532-3544.
[5] Saftig P, Klumperman J. Lysosome biogenesis and lysosomal membrane proteins: trafficking meets function[J]. Nat Rev Mol Cell Biol, 2009, 10(9):623-635.
[6] Boya P. Lysosomal function and dysfunction: mechanism and disease[J]. Antioxid Redox Signal, 2012, 17(5):766-774.
[7] Nielsen R, Christensen EI. Proteinuria and events beyond the slit[J]. Pediatr Nephrol, 2010, 25(5):813-822.
[8] Theilig F, Kriz W, Jerichow T, et al. Abrogation of protein uptake through megalin-deficient proximal tubules does not safeguard against tubulointerstitial injury[J]. J Am Soc Nephrol, 2007, 18(6):1824-1834.
[9] Grimm S, Horlacher M, Catalgol B, et al. Cathepsins D and L reduce the toxicity of advanced glycation end pro-ducts[J]. Free Radic Biol Med, 2012, 52(6): 1011-1023.
[10]胡鹏飞,赖东武,何 红.自噬在晚期糖基化终产物诱导的内皮细胞凋亡中的作用 [J]. 中国病理生理杂志,2012, 28(6):1006-1011.
[11]Tang R, Yang C, Tao JL, et al. Epithelial-mesenchymal transdifferentiation of renal tubular epithelial cells induced by urinary proteins requires the activation of PKC-α and βI isozymes[J]. Cell Biol Int, 2011, 35(9):953-959.
[12]Boya P, Kroemer G. Lysosomal membrane permeabilization in cell death[J]. Oncogene, 2008, 27(50):6434-6451.
[13]Dudeja V, Mujumdar N, Phillips P, et al. Heat shock protein 70 inhibits apoptosis in cancer cells through simultaneous and independent mechanisms[J]. Gastroenterol, 2009, 136(5):1772-1782.
[14]Johansson AC, Appelqvist H, Nilsson C, et al. Regulation of apoptosis-associated lysosomal membrane permeabilization[J]. Apoptosis, 2010, 15(5):527-540.
[15]于春艳,刘 希,张 钰,等. 自噬溶酶体抑制剂氯化铵促进维生素K3诱导人宫颈癌HeLa细胞凋亡的作用 [J]. 中国病理生理杂志,2013, 29(7): 1197-1201.
[16]Repnik U, Stoka V, Turk V, et al. Lysosomes and lysosomal cathepsins in cell death [J]. Biochim Biophys Acta, 2012, 1824(1):22-33.
[17]Caruso-Neves C, Pinheiro AA, Cai H, et al. PKB and megalin determine the survival or death of renal proximal tubule cells [J]. Proc Natl Acad Sci U S A, 2006, 103(49):18810-18815.
[18]Ohse T, Inagi R, Tanaka T, et al. Albumin induces endoplasmic reticulum stress and apoptosis in renal proximal tubular cells[J]. Kidney Int, 2006, 70(8): 1447-1455.
[19]Urahama Y, Ohsaki Y, Fujita Y, et al. Lipid droplet-associated proteins protect renal tubular cells from fatty acid-induced apoptosis[J]. Am J Pathol, 2008, 173(5):1286-1294.
[20]Arici M, Chana R, Lewington A, et al. Stimulation of proximal tubular cell apoptosis by albumin-bound fatty acids mediated by peroxisome proliferator activated receptor-gamma[J]. J Am Soc Nephrol, 2003, 14(1):17-27.
[21]Liu WJ, Luo MN, Tan J, et al. Autophagy activation reduces renal tubular injury induced by urinary proteins[J]. Autophagy, 2014, 10(2):243-256.
[22]Osicka TM, Kiriazis Z, Pratt LM, et al. Ramipril and aminoguanidine restore renal lysosomal processing in streptozotocin diabetic rats[J]. Diabetologia, 2001, 44(2):230-236.
[23]Osicka TM, Houlihan CA, Chan JG, et al. Albuminuria in patients with type 1 diabetes is directly linked to changes in the lysosome-mediated degradation of albumin du-ring renal passage[J]. Diabetes, 2000, 49(9):1579-1584.
[24]Masini M, Bugliani M, Lupi R, et al. Autophagy in human type 2 diabetes pancreatic beta cells[J]. Diabetologia, 2009, 52(6):1083-1086.
[25]Shechter P, Boner G, Rabkin R. Tubular cell protein degradation in early diabetic renal hypertrophy[J]. J Am Soc Nephrol, 1994, 4(8):1582-1587.
[26]Yung S, Cheung KF, Zhang Q, et al. Mediators of inflammation and their effect on resident renal cells: implications in lupus nephritis[J]. Clin Dev Immunol, 2013, 2013: 317682.
[27]刘海燕,陈孝文,梁 东,等.尿毒症毒素对人肾小管上皮细胞外基质的影响及临床意义[J]. 中华急诊医学杂志, 2006, 15(7):620-624.
缺血性琥珀酸聚集通过线粒体ROS控制再灌注损伤
多种疾病尤其是心力衰竭和卒中时,某一器官血供被阻断后血流再通会出现缺血再灌注损伤。对缺血组织再灌注是维系组织生存的必要措施,也会因此通过线粒体产生活性氧簇(ROS)而启动氧化损伤、细胞死亡以及异常的免疫应答过程。尽管已明确缺血再灌注损伤过程中线粒体会产生ROS,但目前普遍认为这是一种非特异性反应。一组英美科学家采用体内代谢产物比较分析方法,出乎意料地发现与缺血再灌注相关的线粒体ROS产生是一条广泛存在的保守代谢通路。他们发现由三羧酸循环介导产生的中间代谢产物琥珀酸的选择性聚集是一个普遍存在于缺血性组织中的代谢标志物,并且是再灌注过程中ROS产生的关键分子。嘌呤核苷酸降解和苹果酸/天冬氨酸穿梭反应的部分逆转造成延胡索酸过多,并反过来促使琥珀酸脱氢酶逆转,从而导致缺血性琥珀酸聚集。再灌注后,聚集的琥珀酸快速地被琥珀酸脱氢酶再氧化,通过逆转线粒体复合体 I 中的电子转运促进过多的ROS生成。体内实验发现用药物抑制缺血性琥珀酸聚集能够有效改善心力衰竭和卒中模型鼠的缺血再灌注损伤。该研究确认了组织对于缺血和再灌注的一种保守代谢反应,从而统一了关于缺血再灌注不同方面的认识,并由此揭示了控制体内ROS产生的新的代谢路径,提示抑制缺血性琥珀酸聚集及再灌注后其氧化产物的生成将成为缺血再灌注损伤疾病的治疗靶点。
Nature, 2014, 515(7527):431-435(麦鸿成)
Effect of urinary proteins and advanced glycosylation end products on lysosomes in renal tubular epithelial cells
DENG Jian-kun, WANG Shu-jun, WU Hong-luan, LUO Mian-na, XU Bi-hua, LIANG Dong, PAN Qing-jun, LIU Hua-feng, LIU Wei-jing
(InstituteofNephrology,TheAffiliatedHospitalofGuangdongMedicalCollege,Zhanjiang524001,China.E-mail:liuweijing-1977@hotmail.com)
AIM: To investigate the effects of pathological products, urinary proteins and advanced glycosylation end products (AGE) produced in the progression of chronic kidney disease (CKD), on the structure and function of lysosomes in renal tubular epithelial cells (TECs), and try to find a novel approach for preventing or delaying CKD. METHODS: The renal specimens of the untreated patients with minimal change nephrotic syndrome (MCNS), diabetic nephropathy (DN) or normal kidney were collected. The expression of lysosomal-associated membrane protein 1 (LAMP1) and cathepsin B (CB) was studied in TECs by indirect immunofluorescent staining. Human renal tubular epithelial cell line HK-2 was incubated with 8 g/L urinary proteins or 100 mg/L AGE. The expression of LAMP1 and CB was investigated by indirect immunofluorescence and the activity of CB and cathepsin L (CL) was measured by biochemical and enzymatic assays.The degradation of DQ-ovalbumin was also determined. RESULTS: The lysosomal membrane permeabilization occurred in the TECs of MCNS and DN patients. After treatment with urinary proteins or AGE-BSA, the lysosomal membrane permeabilization of the HK-2 cells was increased. The activity of CB and CL and degradation of DQ-ovalbumin were decreased as compared with normal control group. CONCLUSION: The digestive function of lysosome was decreased and lysosomal membrane permeabilization occurred in the TECs exposed to urinary proteins and AGE, which might be a key factor to induce the tubulointerstitial fibrosis.
Tubular epithelial cells; Lysosome; Urinary proteins; Advanced glycosylation end products
1000- 4718(2015)03- 0505- 06
2014- 09- 18
2014- 12- 09
国家自然科学基金资助项目(No. 81270798);广东省自然科学基金资助项目(No. S2012040006276);广东省医学科研基金资助项目(No. A2014480);湛江市科技攻关计划(No. 2013B01056);广东医学科研基金(No,m2011009)
△通讯作者 Tel: 0759-2387164; E-mail: liuweijing-1977@hotmail.com
▲并列第1作者
R363.2+1
A
10.3969/j.issn.1000- 4718.2015.03.021
猜你喜欢
河南科技(2022年6期)2022-04-22
复旦学报(医学版)(2021年5期)2021-10-13
生物化工(2021年2期)2021-01-19
健康之家(2020年15期)2020-05-08
读与写(2019年35期)2019-11-05
中国医学创新(2019年9期)2019-08-19
癌症进展(2018年11期)2018-12-30
现代职业教育·高职高专(2018年7期)2018-05-14
医学研究杂志(2015年7期)2015-06-22
医学研究杂志(2015年12期)2015-06-10
