基于CASTEP软件对非化学计量比TiC的结构研究
2016-01-27 04:36贺战文
武汉轻工大学学报 2015年4期
贺战文,张 琰
(1.武汉轻工大学 机械工程学院,湖北 武汉 430023,2.武昌工学院 信息工程学院,湖北 武汉 430065)
基于CASTEP软件对非化学计量比TiC的结构研究
贺战文1,张琰2
(1.武汉轻工大学 机械工程学院,湖北 武汉 430023,2.武昌工学院 信息工程学院,湖北 武汉 430065)
摘要:TiC因其具有众多优异的物理及化学性能而在很多领域引起人们的广泛关注,并获得广泛应用。然而,由于其熔点高、烧结困难,在某种程度上又限制了它的发展应用。本文是基于密度泛函理论,利用第一性原理赝势方法结合Materials Studio中的CASTEP计算模块,先构建出正常价态TiC晶胞模型并进行结构优化运算,得到其能量与态密度的相关数据。再构建非化学计量比TiC晶胞模型,优化结构并计算晶格常数,得到其能带图和态密度图。最终对比分析三种不同结构的非化学计量比TiC的能带图和态密度图与正常价态TiC的态密度图和能带图,得到非化学计量比的存在对TiC性质的影响,并与文献报道的实验结果相吻合。
关键词:非化学计量比;碳化钛;机械合金化;第一性原理
1引言
近代晶体结构的理论与实验研究结果表明,具有化学计量比与非化学计量比的化合物都是普遍存在的,且非化学计量比化合物的存在更为普遍。非化学计量比化合物往往由于各种缺陷的存在,在光、电、声、磁、力、热等方面呈现特殊的性质,使得非化学计量比化合物成为一种很好的功能材料。因此对非化学计量比化合物的研究具有极大的意义。
非化学计量比化合物(Non-stoichiometric Compound)是指偏离化学式的化合物,化合物中化学成分与晶体结构中的原子没有一个固定的比例,所以它的化合价并不是整数[1]。在化合物的晶胞中的空位对性能有十分明显的影响,从而非化学计量化合物也显示出了不同一般的性能,而有很多典型的非化学计量化合物,因为在硬度方面有良好的表现引起人们的关注。这些物质的具体细化分类对材料科学的研究以及发展有十分重大的含义。根据近现代的实验研究成果,人们发现非化学计量比化合物在科研以及现实生活中的意义和用处越来越大。而且因为这些化合物独特的内部分子排列结构,使得由这些化合物组成的物质在很多方面都有优异的性能,但是非化学计量化合物的形成必须满足一定的条件即:0.41< RX/RM<0.59,其中RM与RX分别表示金属原子的原子半径和非金属原子的原子半径[2]。由于晶胞中空位的作用,使得非化学计量化合物在很多方面表现出优异的性能。金属碳化物、氮化物是常见的非化学计量化合物,在硬质结构材料性能优良,因而而受到广泛关注。其中过渡金属与碳、氧、氮、硫、硼、氢的立方化合物和一些三元化合物都是典型的非化学计量化合物。
表1为一些是金属和非金属的原子X(X=N,C)与金属原子(用M表示)的原子半径数值的比值。
表1部分金属原子半径以及非金属原子X(X=C,N)与金属原子M的原子半径比
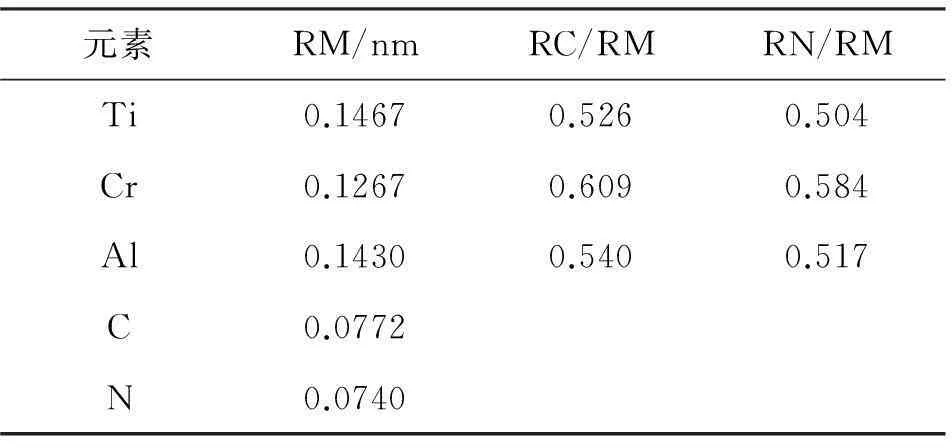
元素RM/nmRC/RMRN/RMTi0.14670.5260.504Cr0.12670.6090.584Al0.14300.5400.517C0.0772N0.0740
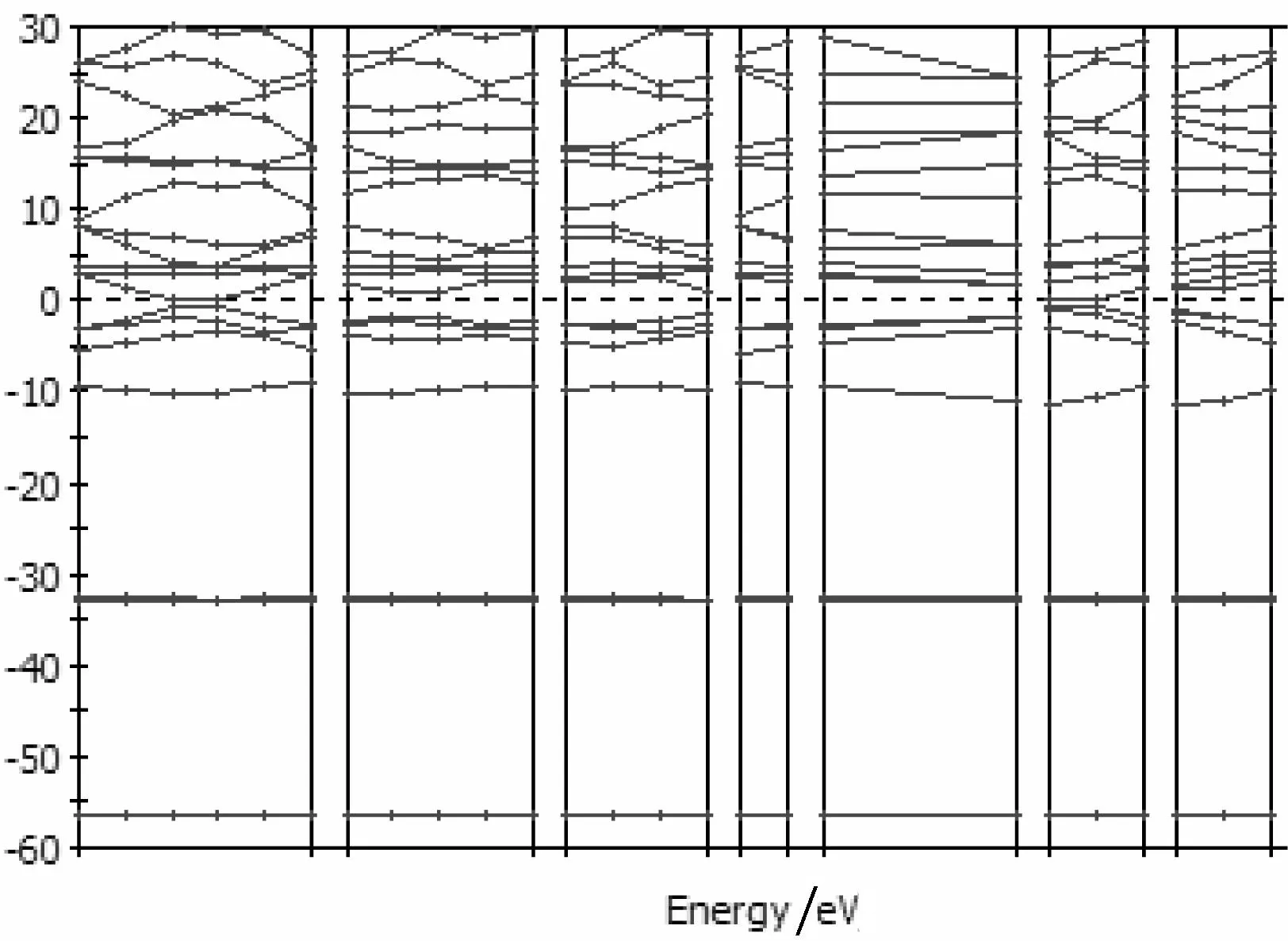
由表1可知,0.41 通过基于密度泛函理论的第一性原理,利用材料学分析计算软件Materials Studio 中的CASTEP模块对非化学计量比TiC和化学计量比TiC的晶体结构进行了模拟构建,并优化其晶胞结构,通过运算得出其态密度曲线和能带图。在对比了相关的实验数据[3]之后,发现与计算结果相吻合,证实计算结果可靠。说明通过形成非化学计量比碳化钛的确可以提高碳化钛的韧性,降低了烧结温度。 2实验方案 实验中主要利用CASTEP软件,对正常化学计量比的TiC和非化学计量比TiC进行带结构和态密度这两项计算。在利用CASTEP对非化学计量比TiC进行能量和态密度计算的过程中难点主要在于非化学计量比TiC晶胞模型的构建。这个过程需要先构建正常化学计量比TiC的晶胞模型,然后根据所要计算的非化学计量比TiC中Ti与C的摩尔比再从已构建好的模型中拿掉相应的原子,使其达到所需要的原子摩尔比,然后对结构进行优化,再设置计算参数开始计算。通过对Ti与C的摩尔比为1:0.5的TiC0.5非化学计量比化合物与正常化学计量比TiC相比,计算得到的能带结构图及态密度图进行分析(见图1—图6),并进行比较,根据能带结构图及态密度图中所呈现的数据推出结论。 3实验结果与讨论 构建一个晶格的结构,需知道该晶体的空间群、晶格参数、以及晶体里面的原子坐标。TiC是面心立方NaCl结构,空间群为Fm3m代号为225,晶体里面一个原胞有两个原子,Ti原子和C原子的分数坐标分别为(0,0,0)和(1/2,1/2,1/2),本文采用的是初基原胞计算,它的大小为20.313,实验研究的计算结果见图1—图6。 图1 化学计量比TiC结构 图2 非化学计量比TiC0.5结构 图3 TiC能带图 图4 TiC0.5能带图 图6 TiC0.5态密度图 图3和图4是TiC与 TiC0.5的能带图,能带是用来说明在晶体中电子的运动情况,价电带一般是值在0K时,固体材料里面的电子所含有的最高的能量。在导带中,电子的能量的范围一般高于价带,从而在传导带中的电子都可以在材料之外的电场中发生运动从而形成电流。从正常价态的TiC和非化学计量比的TiC0.5能带图中可以看出,在-2eV附近能带图中能带的密集度不断减小。说明当化合物中C的含量降低时,Ti-C键的浓度降低,而Ti-C键具有很强的键能,最终导致TiC的烧结温度降低,这与实际实验得到的结果相吻合。 图5和图6是态密度图。TiC0.5态密度图是软件所模拟的非化学计量比TiC0.5态密度图。通过对图5和图6的分析,可以推测出所测物质中的化学元素和他们的价态。从TiC和TiC0.5的态密度图中可以看出随C含量的降低,-3 eV附近成键峰逐渐变小,说明Ti-C键的浓度逐渐降低。在Femi能级附近,出现空位峰,随空位浓度的增加,峰逐渐变大。由此可以看出,随着空位浓度的增加Ti-C的金属性也有所增加,具体表现为强度硬度基本不变,而韧性增强,与已知事实一致。 在分析能带图和态密度图的时候发现如下规律,能带越平稳的地方,态密度图的峰值越大,能带跨度越大,则在态密度图中,图像的峰跨过得能量范围越宽广,相应峰值的变化也越平稳。总之,能带越平,态密度图越尖锐,能带跨度越大,态密度越平滑,离域性越强。 利用第一性原理,通过CASTEP软件计算可以得出TiC的体积模量为321GPa,在C所占百分比变小的同时,它的体模量也在变小,也就是说硬度也在降低。而非化学计量比的TiC0.5体模量为153GPa。这个数据的变小主要是因为在TiC0.5中存在着空位,由于这些空位的存在使得整个材料内 部的能量变小,与此同时,空位也会让材料中产生出键能不强的Ti-Ti键,而这种脆弱的金属键让TiC在保持硬度和强度的同时增加了足够的韧性同时还可以极大的降低TiC的烧结温度。 4结论 笔者利用基于密度泛函理论的第一性原理赝势法,通过理论计算在结合文献[3]的实验数据,对非化学计量比TiCx的结构性能进行研究,得出如下结论。 (1) 非化学计量比TiCx在理论上可以形成,并且在常温下能保持稳定的状态。 (2) TiCx的能量处于-56 eV —36 eV之间,态密度在-2eV的地方达到峰值。 (3) 随着非化学计量比的TiCx形成可以有效的增加韧性并保持强度硬度不降低,同时也可以降低TiC的烧结温度。 参考文献: [1]牛明勤,范莹莹.一种非化学计量钛氧化物的制备和表征[J].同济大学学报(自然科学版);2003(05). [2]Liu L M, Liu H, Ye F, et al. Microstucture and Mechanical Properties of the Spark Plasma Sintered Ta2C Ceramics[J]. Ceramics International, 2012, 39: 4707-4713. [3]孙金峰. MA制备非化学计量比TiCx和TiNx及其烧结特性的研究[D]. 秦皇岛:燕山大学, 2009. Study of the structure of the nonstoichiometric of TiC based on castep software HEZhan-wen,ZHANGYan (1.School of Mechanical Engineering, Wuhan Polytechnic University, Wuhan 430023,China, 2.College of Information Science and Engineering,Wuchang Institute of Technology, Wuhan 430065,China) Abstract:Because of its many excellent physical and chemical properties TiC caused widespread concern in many areas, and is widely available. However, its high melting point, and firing difficult, to some extent limited its development and application. Related data presented herein is based on density functional theory, and the use of first-principles pseudopotential method combines the Materials Studio CASTEP calculation module, first build a normal valence TiC lattice model and structure optimization calculation to obtain the energy and density of states . Then the construction of non-stoichiometric TiC lattice model, optimize the structure and lattice constants calculation to obtain its energy band diagram and the density of states. Comparison of the final non-stoichiometric ratio of three different structures TiC band diagram and the density of states and valence TiC normal density of states and energy band diagram can get the effect of the presence of non-stoichiometric ratio on TiC in nature and with the results coincide with litterature reports. Key words:nonstoichiometric compound; TiCx; mechanical alloying;the first principle DOI:10.3969/j.issn.2095-7386.2015.04.009 10.3969/j.issn.2095-7386.2015.04.008 文章编号:2095-7386(2015)04-0031-04 2095-7386(2015)04-0027-04 通信作者:张家凡(1963-),男,教授,E-mail: jfz@whpu.edu.cn. 管庶安(1956-),男,教授,E-mail:1592547368@qq.com. 作者简介:张媛媛(1990-),女,硕士研究生,E-mail:413829102@qq.com. 戴立(1990-),男,硕士研究生,E-mail:liiadv5@163.com. 收稿日期:2015-06-18.修回日期:2015-09-22. 2015-06-09. 中图分类号:TG 732 文献标识码:A