
二元醇混合物汽液相平衡的GEMC模拟与微观结构分析
2016-09-20 09:14李洪李东洋李鑫钢高鑫
化工进展 2016年9期
李洪,李东洋,李鑫钢,3,高鑫
(1天津大学化工学院,天津 300072;2精馏技术国家工程研究中心,天津 300072;3天津化学化工协同创新中心,天津 300072)
研究开发
二元醇混合物汽液相平衡的GEMC模拟与微观结构分析
李洪1,2,李东洋1,2,李鑫钢1,2,3,高鑫1,2
(1天津大学化工学院,天津 300072;2精馏技术国家工程研究中心,天津 300072;3天津化学化工协同创新中心,天津 300072)
采用Gibbs蒙特卡罗模拟方法和OPLS-AA力场计算了乙二醇(EG)/1,2-丁二醇(1,2-BDO)、乙二醇/1,3-丁二醇(1,3-BDO)、乙二醇/1,4-丁二醇(1,4-BDO)三组二元醇混合物的汽液相平衡数据,通过与实验数据及Wilson状态方程计算数据的比较验证了方法和力场对体系汽液相平衡预测的适用性。对混合体系饱和液相的径向分布函数分析可知,各二醇间存在与羟基官能团位置近似无关的强氢键作用和作用范围不同的范德华作用。经体系 O-O平均数目的统计及氢键缔合比例分析发现,随乙二醇组成增大,3种体系的微观结构由主要包含较大的BDO-BDO、BDO-EG缔合结构(>6)向包含较小的EG-BDO、EG-EG缔合结构(2~4)转变,且缔合比例下降。Molclus+Gaussian09对小分子EG、1,2-BDO缔合结构进行能量优化与构型搜索,由此提出了二醇缔合比例随EG组成变化的可能原因:1,2-BDO间缔合结构稳定于其他缔合结构,随缔合分子数增大,各缔合结构稳定性增加,但EG自缔成大分子结构的稳定性与其和BDO缔合成小分子结构稳定性差异减小。
Gibbs蒙特卡洛模拟;乙二醇/1,2-丁二醇;汽液相平衡;微观结构
乙二醇(EG),又被称为甘醇,是一种需求量很大的石油化工原料。随着石油资源的逐渐短缺,采用非石油路线生产乙二醇的工艺具有战略性意义,但非石油路线制备乙二醇中出现了 1,2-丁二醇(1,2-BDO)、1,3-丙二醇(1,3-PDO)和1,4-丁二醇(1,4-BDO)等副产物,所以提纯乙二醇显得至关重要。其中乙二醇/1,2-丁二醇的分离是煤基或生物基生产高纯度乙二醇的关键技术难题之一[1]。相平衡数据是混合物分离工程计算和设计的依据,目前对于乙二醇/1,2-丁二醇混合物的汽液相平衡数据已有实验报道[2],但相关研究并未对体系微观构型的差异性进行研究和分析。关于一元醇混合物的微观结构和氢键缔合的研究已有相关报道[3-4],但还不能够解释不同二元醇混合物的构型特点及其形成原因。分子模拟方法作为实验法的有效补充[5],可以被用来研究以上问题,其中Gibbs系综蒙特卡罗方法对混合物汽液相平衡的预测的准确性已有报道[6-7]。
本文针对以上问题,采用Gibbs蒙特卡罗方法、OPLS-AA力场[8]对 EG/1,2-BDO、EG/1,3-BDO、EG/1,4-BDO等二元醇混合物的汽液相平衡性质进行计算,经与实验结果或Wilson状态方程计算数据进行对比来确定方法和力场的适用性,通过径向分布函数分析总结,试图解释各二醇混合物微观结构特点的差异性。同时,基于量化方法对不同异种分子缔合构型进行搜索,以求找到形成以上差异性的可能原因。
1 力场模型
本研究采用的力场模型是OPLS-AA力场,它是一种适用范围较广的全原子力场。力场中的范德华作用采用Lennard-Jones 12-6(LJ 12-6)势能模型来计算,长程静电作用则通过静电势能模型来计算,相关参数rij、εij、σij、qij和qj分别代表了i和j距离、势阱深度、碰撞直径及原子i和j的电荷数,见式(1)。其中 LJ势能模型中参数εij、σij通常采用Geometric混合规则由εi、εj、σi、σj计算获得,如式(2)和(3)所示,以上模拟所用的非键参数见表1。
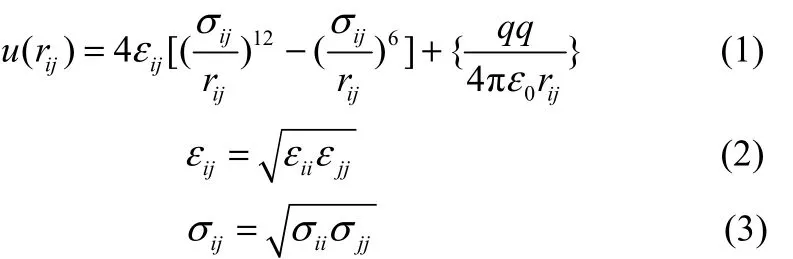
相关的键参数见表2~表4,其中键长采用固定键长形式如式(4),键角采用谐振角弯曲形式如式(5),二面角采用余弦扭转角形式如式(6)。

2 模拟方法
应用Towhee软件[10]调用OPLS-AA力场在常压下对总分子数为450的乙二醇/1,2-丁二醇混合物的汽液相平衡进行了模拟计算。体系初始构型由随机数发生器随机产生,并采用周期性边界条件。范德华势能采用12~14Å半径截断,并对长程作用力施加尾部矫正,静电作用采用Ewald加合法和锡箔边界条件(κ×L=5,Kmax=5)处理。随机Monte Carlo运动包括0.003的体积涨落,余下分别平均分配给分子平动、分子转动及分子在盒间的交换和偏倚运动。体系均通过80000循环步长达到预平衡,另经过 60000循环步长进行平衡。最后,使用MOLCLUS[11]和 GAUSSIAN[12]进行构型搜索时采用的方法和基组是M062/6-31g(d,p)。
3 结果与讨论
3.1 汽液相平衡
将EG/1,2-BDO混合物汽液相平衡的计算结果与已报道的实验数据进行了比较,结果如图1、表5所示。由表5可见,液相组成计算偏差小于6%,气相组成计算偏差小于7%,表明GEMC方法、OPLS-AA力场适用于计算该体系的汽液相平衡性质。同样的,将EG/1,3-BDO和EG/1,4-BDO汽液相平衡的模拟数据与 Wilson状态方程计算数据进行比较,结果见表6、表7、图2、图3。经比较可知,前者液相组成偏差小于10%,气相偏差小于5%,后者液相组成偏差小于11%,气相偏差小于10%。虽然两者相对EG/1,2-BDO相平衡组成计算偏差较大,但能较准确地描述汽液相平衡曲线的变化趋势。

表1 范德华作用和静电作用势能参数[8-9]

表2 各组分键长参数[10]

表3 各组分键角弯曲参数[10]
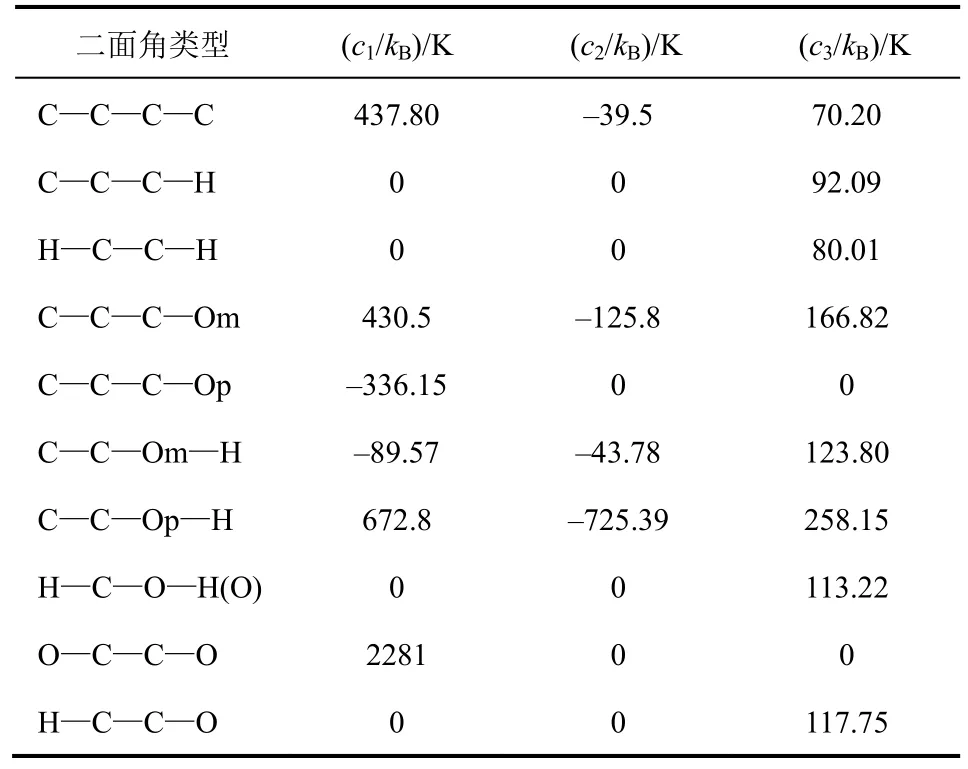
表4 各组分二面角扭曲参数[10]
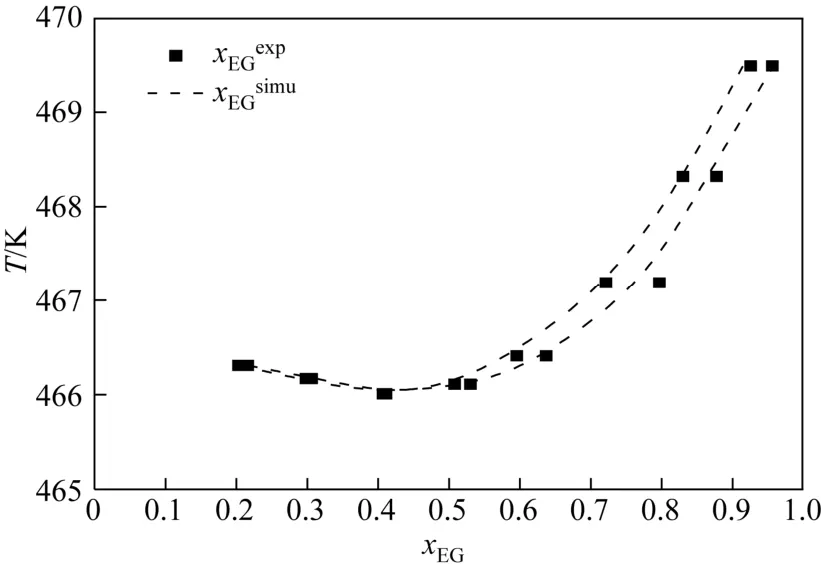
图1 乙二醇/1,2-丁二醇混合物汽液相平衡曲线
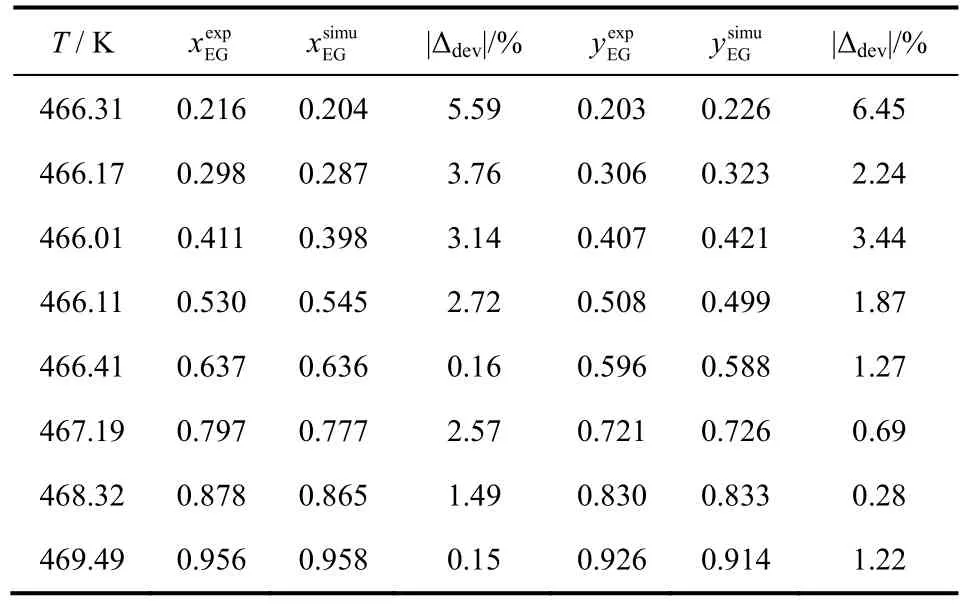
表5 OPLS-AA力场下乙二醇/1,2-丁二醇混合物汽液相平衡数据
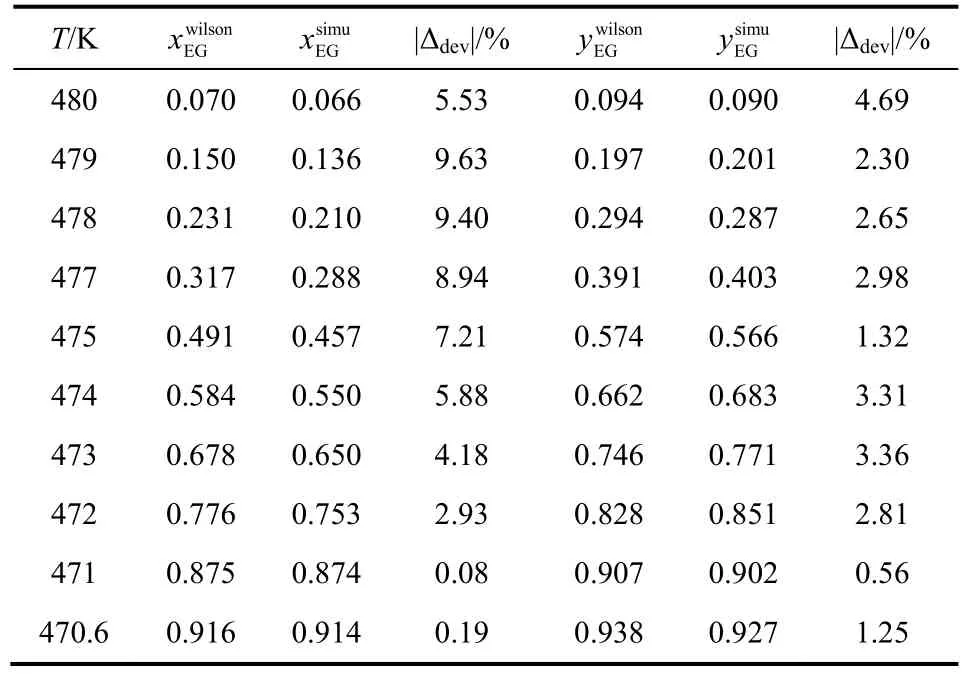
表6 OPLS-AA力场下乙二醇/1,3-丁二醇混合物汽液相平衡数据
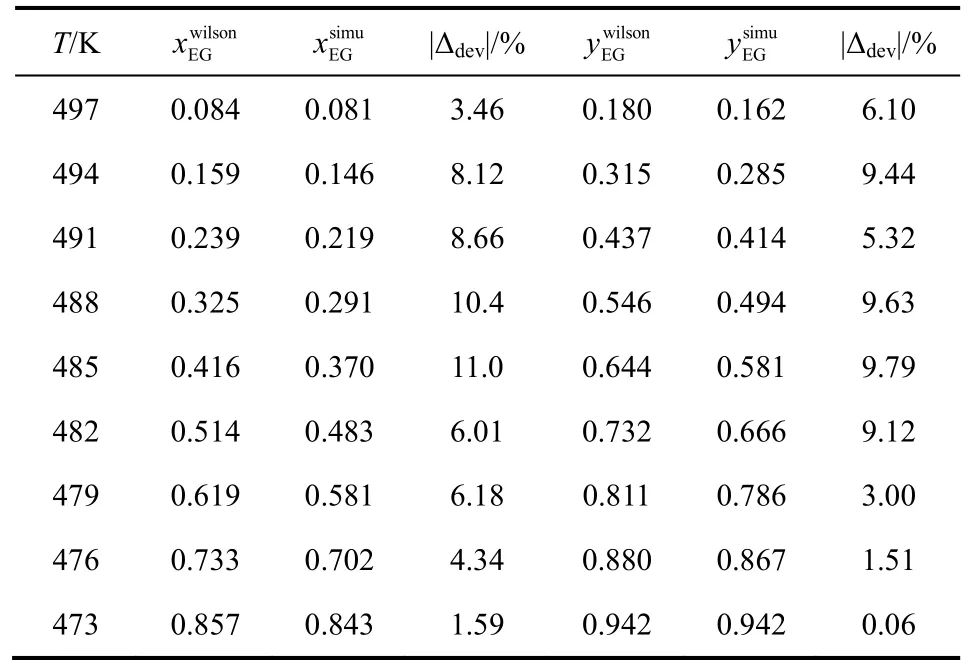
表7 OPLS-AA力场下乙二醇/1,4-丁二醇混合物汽液相平衡数据

图2 乙二醇/1,3-丁二醇混合物汽液相平衡曲线

图3 乙二醇/1,4-丁二醇混合物汽液相平衡曲线
3.2 径向分布函数分析
径向分布函数(radial distribution functions,RDFs)指在指定粒子一定距离内找到另一粒子的概率密度,通常用来研究距离指定原子(或分子)的其他粒子在空间的分布概率,是分析物质微观结构特性的常用工具[13]。
为研究以上各混合物微观结构的差异性,文章分别描述并分析了 EG/1,2-BDO、EG/1,3-BDO、EG/1,4-BDO 饱和液相各组分氧原子间径向分布函数(go-o)。以近似相等组成的平衡饱和液相(xEG≈0.50)作为分析对象,将各径向分布函数描述如图4~图6。由图4~图6可见,EG/1,2-BDO混合物中各 go-o第一峰峰值约为1.6,EG/1,3-BDO混合物中go-o第一峰峰值在1.3~1.5,EG/1,4-BDO混合物中 go-o第一峰峰值在 1.3~1.7,且均出现在约2.8Å处。说明3种混合体系中存在大量异种、同种分子因氢键缔合结构,但不同体系中氢键缔合结构的组成比例可能有所差异,要进一步说明这种差异,还需对混合物中的氢键缔合比例进一步分析。值得注意的是,1,4-BDO分子间go-o第一峰峰值相对偏小,考虑到其官能团分布在分子两端、链长相对本研究中其他二醇分子较长,因此缔合产生的长链状构型稳定性较差。此外,3种混合体系中 go-o第二峰峰值的出现位置有所差异,其中EG分子间go-o第二峰峰值出现在约4.8Å处,EG与BDO分子间go-o第二峰峰值出现在约5.4Å处,而BDO分子间go-o第二峰峰值出现在约5.8Å处,这说明3种混合体系中均存在一定的弱相互作用,但不同分子结构对其作用范围造成了影响,即链长最短的EG在相对较短距离处产生了弱相互作用,而链长最长的BDO则在相对较远处才产生,EG与BDO的弱相互作用出现在两者之间。综上,经径向分布函数分析可知,3种二元醇混合物中存在大量与官能团所在位置无紧密关系的氢键缔合作用,且不同二元醇间将产生不同距离的弱相互作用。
本文还对不同体系异种分子间径向分布函数go-o进行了分析,图7~图9分别描述了在不同EG组成下(图7,xEG=0.20、0.51、0.93;图 8,xEG=0.14、0.51、0.81;图9,xEG=0.15、0.49、0.84),EG与1,2-BDO醇、1,3-BDO、1,4-BDO的go-o变化规律。如图所示,3个体系中 go-o第一峰峰值变化均较小,在1.5~1.7之间,产生于约2.8Å处,同时第二峰峰值在1.1~1.2之间,均发生在约5.4Å处。以上说明随着EG组成的变化,异种二醇分子间因氢键发生的缔合作用无较大变化,且EG与官能团位置不同的 BDO之间的氢键缔合作用也未发生较大变化,即 BDO官能团位置的不同并未对两者之间的强氢键作用造成影响。
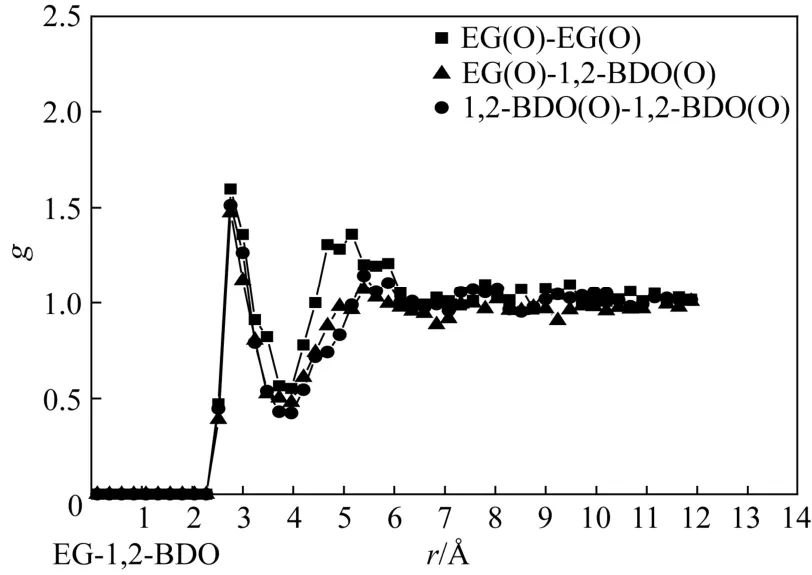
图4 液相乙二醇组成为0.54时,乙二醇、1,2-丁二醇各羟基间径向分布函数

图5 液相乙二醇组成为0.51时,乙二醇、1,3-丁二醇各羟基间径向分布函数
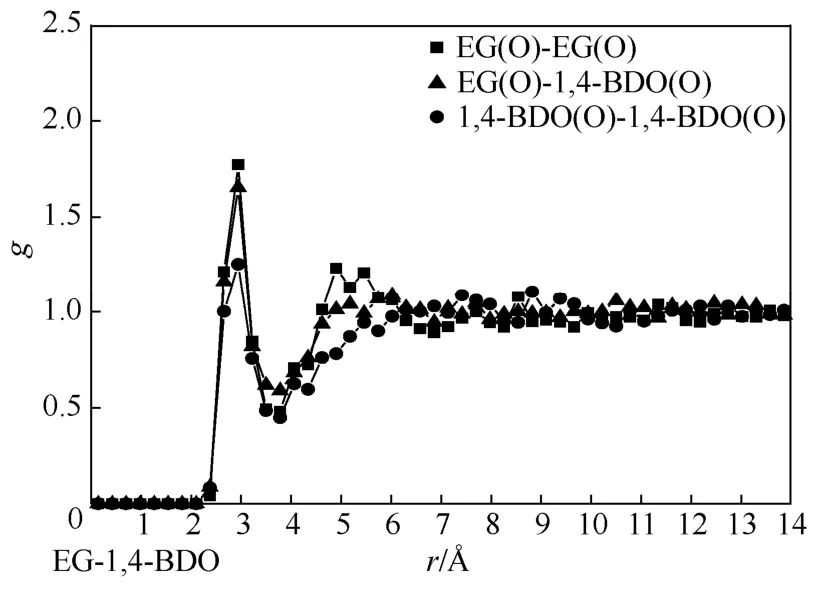
图6 液相乙二醇组成为0.49时,乙二醇、1,4-丁二醇各羟基间径向分布函数
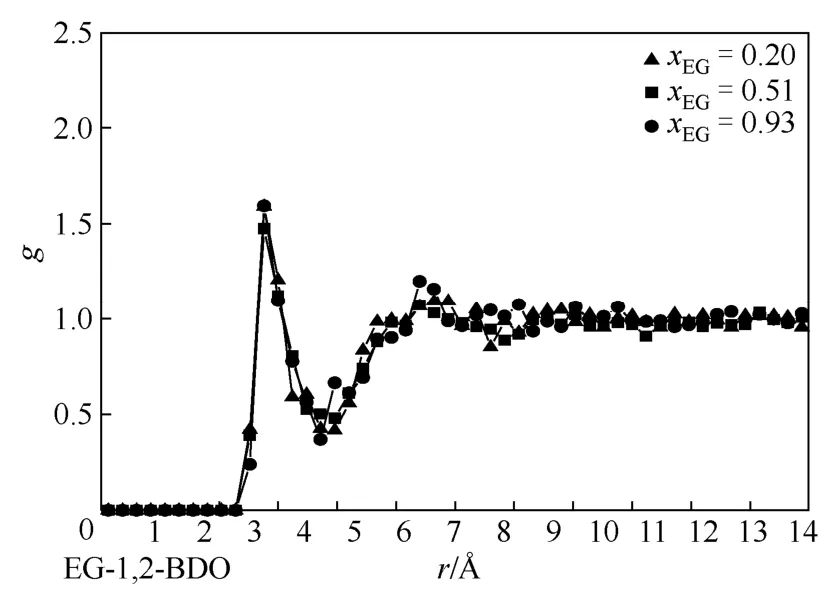
图7 不同饱和液相乙二醇组成下,乙二醇与1,2-丁二醇羟基间径向分布函数
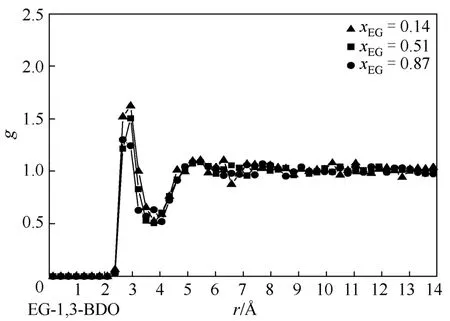
图8 不同饱和液相乙二醇组成下,乙二醇与1,3-丁二醇羟基间径向分布函数

图9 不同饱和液相乙二醇组成下,乙二醇与1,4-丁二醇羟基间径向分布函数
3.3 氢键缔合分析
为了进一步比较混合物中不同二元醇由于氢键缔合比例,本文对混合物中O-O原子对平均数目进行了计算统计。通过积分相应的RDF曲线并乘以其相对原子数密度,可得到不同EG组成下O-O原子对的平均数目,它可以用来描述各分子因氢键缔合比例的趋势,孙淮等[14]曾用此方法成功描述过EG/H2O混合物中O-O原子对平均数目的变化,本研究对3种混合物O-O的平均数目统计如表8所示。比较不同混合物随饱和液相组成变化的O-O原子对平均数目可知,当 EG组成升高时,OEG-OEG的数目都逐渐增大且变化范围较广,而OBDO-OBDO数目逐渐减小,变化范围也较广,相比之下,OEG-OBDO数目先减小后增大,且变化范围较窄。研究结果表明,在EG组成较小时,EG更倾向于与BDO进行缔合,随着EG数目增多,EG自身缔合数目增加,同时EG自身或与BDO产生缔合的比例将趋于近似相等,而BDO由自缔合则逐渐转变为与EG之间的缔合。在EG组成约0.5时,虽然O-O原子对数目关系是:NEG-EG>NEG-BDO>NBDO-BDO,但相差均较小,说明此时混合物液相存在较小的负偏差,实现了相对其他EG组成时更加均匀的缔合,因而形成了OEG-OBDO的极小值。综上,表8描述了各二醇体系O-O原子对数目的变化情况,反映了其缔合结构变化规律的一致性,这对正确理解二醇混合物微观结构的共同特点有一定的指导意义,同时也间接证明OPLS力场和GEMC方法能够实现对二醇混合物饱和液相中氢键缔合比例的预测。
以上 O-O原子对的统计对混合物中氢键缔合趋势有一定的预测作用,然而要更进一步确定其氢键缔合结构的比例,则需要使用严格的几何规则对其进行搜索与统计。采用 Luzar和 Chandler提
出的经典几何判据(RO1O3≤3.3Å,RO1H4≤2.4,θH4-O3…O1≤30°)[15],用以计算各二醇混合物饱和液相中形成氢键分子数目的比例,SAIZ等[16]成功用此统计规则对不同力场模型下的氢键结构进行了统计。表9描述了3种二醇混合物中不同组成下强氢键缔合分子数目占饱和液相总分子数的比例。由表9可知,3种体系中强氢键缔合比例随xEG的变化产生了巨大的差异,在xEG较小时体系中出现了大量分子数目大于6的缔合结构(EG-1,2-BDO∶0.31;EG-1,3-BDO∶0.23;EG-1,4-BDO∶0.23),随着xEG的升高,体系中此类多元缔合结构的数目快速下降,转变为以2~3分子缔合为主。相较而言,在xEG约0.5时,体系中分子缔合比例分布稍显平均,各有10%~15%的2~6元分子发生缔合。此外,3种混合物中缔合分子比例均随xEG增大而逐渐减小,当xEG最大时体系中约有 40%~50%的分子未发生强氢键缔合。综上,在xEG较大时EG更倾向于与自身或BDO形成短链(2~3分子)缔合构型,而在xEG较小时,BDO更倾向于与自身或EG缔合成长链(>4分子)构型,这可能与两类分子间形成不同缔合结构的稳定性有关。值得注意的是,以上结论与表8所得通过O-O原子对变化对氢键缔合的预测结果相一致,从而也验证了其方法的正确性。

表8 O-O原子对平均数目
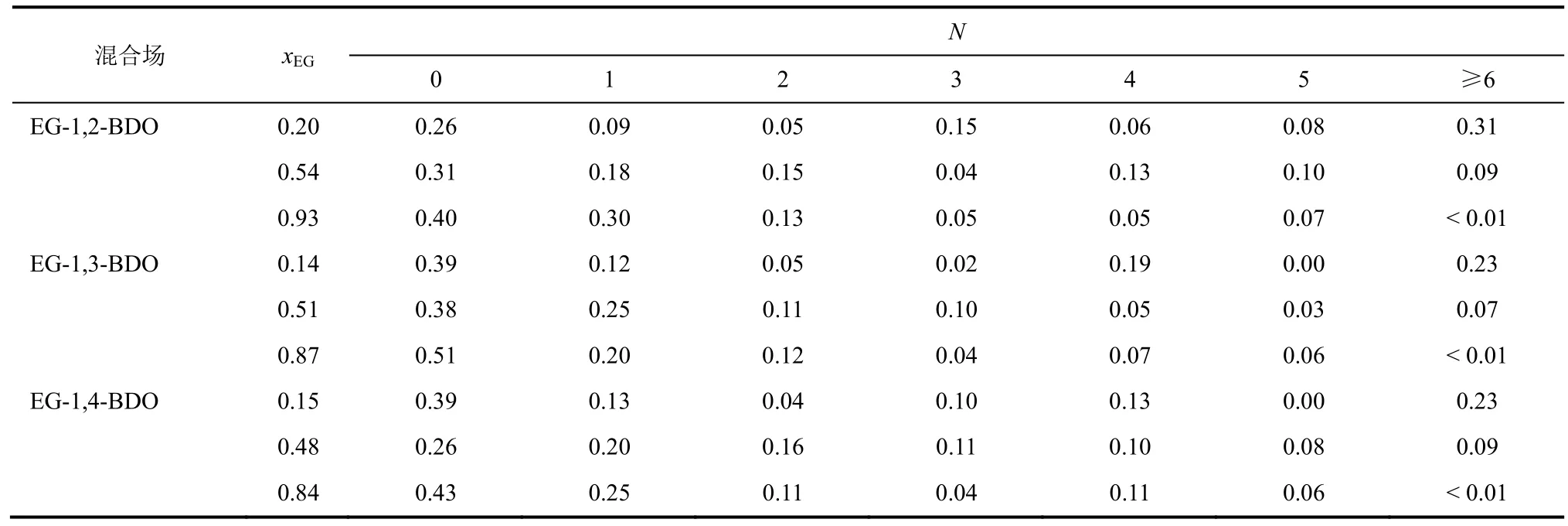
表9 含N个氢键缔合结构的分子占混合物平衡液相总分子数比例
本研究还尝试从能量角度进一步解释以上径向分布函数分析及氢键缔合统计所得结论,因此通过Molclus+Gaussian09对混合物各分子间缔合结构进行了搜索与筛选。以EG/1,2-BDO混合物为例,经Molclus搜索可得各缔合构型的最优结构,其能量排列如表10所示。需要指明的是,计算机对构型搜索的耗时与分子数及其大小成正比,为了方便研究,文章仅对其中的 2~4元氢键缔合结构进行了搜索及能量排序。由表10可见,随缔合分子数的增多,缔合构型的稳定性增强,且对同等分子数形成的缔合结构其稳定性关系是:BDO(BDO)>EG(BDO)>EG(EG)。这即是说,当饱和液相中xEG较小时,EG更倾向于与BDO分子缔合成稳定结构,但随着EG组成增多,虽然可自缔成大分子构型,稳定性却仅近似或稍大于它与 BDO之间形成的缔合结构,使得EG自缔成多分子构型的概率与其和BDO之间形成多分子构型的概率相差不大,即是说在EG组成较大时,EG并不具备自缔成多分子构型(N≥4)的条件。这解释了为何在xEG较大时体系中2~3元缔合结构占重要的比例,此时虽然EG分子较多,但其缔合结构的稳定性不如与 BDO缔合的强。也解释了为何在xEG较小时,BDO可以与BDO及EG形成较大分子的缔合结构,此时BDO分子自缔合较易形成相对稳定的结构。以上与从表8中O-O原子对平均数目分析所得结论和表9氢键缔合分析结果相一致,揭示了导致EG/1,2-BDO中各氢键缔合结构变化趋势的可能原因,同时也说明通过Molclus+Gaussian能够对该类二醇混合物中缔合结构的变化进行简单预测。

表10 EG/1.2-BDO各氢键缔合构型能量
4 结 论
本文采用GEMC模拟方法计算了EG/1,2-BDO、EG/1,3-BDO、EG/1,4-BDO 3种混合物的汽液相平衡性质及微观结构,得到的主要结论如下。
(1)验证了GEMC方法和OPLS-AA力场对3种混合物的汽液相平衡性质计算的适用性。
(2)混合物体系中各二醇间均存在较强的氢键作用和一定的范德华作用,且强氢键作用与二醇羟基位置基本无关,范德华力则因不同体系而作用在不同范围。对O-O原子对数目的统计较好地实现了对混合物中不同二醇间氢键缔合比例随xEG变化的预测,使用几何规则对不同体系中强氢键结构的统计验证了以上预测的正确性。
(3)xEG较小时,混合体系中以多元分子缔合(>6)为主,BDO倾向与自身或EG形成大分子缔合团簇结构;xEG约0.5时,体系内缔合比例较为平均,且EG-EG、BDO-BDO及EG-BDO间较易形成2~6分子缔合结构;xEG较大时,EG倾向于与自身或BDO缔合成小分子团簇结构(2~4)。随着xEG变大,饱和液相中分子缔合比例下降。
(4)通过Molclus+Gaussian对EG-1,2BDO小分子团簇结构进行优化搜索,揭示了 EG/1,2-BDO中氢键缔合结构变化趋势的可能原因,即 BDO自身缔合结构稳定性大于其他等分子缔合结构,且随缔合分子数增加,各缔合结构稳定性增加,但小分子BDO-EG缔合结构(N-1)与大分子EG-EG缔合结构(N)稳定性的差异减小,使EG即使在高浓度下也不具备形成大分子缔合(N≥4)的条件。同时,验证了Molclus+Gaussian可以实现对混合物中缔合结构变化规律的简单预测。
符 号 说 明

[1]周张锋,李兆基,潘鹏斌,等.煤制乙二醇技术进展[J].化工进展,2010,29(11):2003-2009.
[2]朱连天,阎建民,肖文德.乙二醇-1,2-丁二醇二元体系汽液平衡数据的测定及关联[J].化学工程,2012,40(7):34-37.
[3]蓝蓉,李浩然,韩世钧.DFT和热力学研究氢键协同效应及对关联1H NMR的影响[J].化学学报,2005(14):55-59.
[4]蓝蓉.含醇缔合体系的理论,实验和分子模拟研究[D].杭州:浙江大学,2005.
[5]THEODOROU D N.Progress and outlook in Monte Carlo simulations[J].Industrial & Engineering Chemistry Research,2010,49(7):3047-3058.
[6]王秀丽.Gibbs 系综 MonteCarlo 方法研究流体相平衡[D].天津:天津大学,2003.
[7]PANAGIOTOPOULOS A Z.Direct determination of phase coexistence properties of fluids by Monte Carlo simulation in a new ensemble[J].Molecular Physics,1987,61(4):813-826.
[8]JORGENSEN W L,MAXWELL D S,Tirado-Rives J.Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids[J].Journal of the American Chemical Society 1996,118:11225-11236.
[9]TAYLOR R S,SHELDS R L.Molecular-dynamics simulations of the ethanol liquid-vapor interface[J].The Journal of Chemical Physics,2003,119(23):12569-12576.
[10]MARTIN M.MCCCS Towhee 7.0.1[CP/OL].http://towhee sourceforge net,2008.
[11]TIAN Lu.Molculs program[CP/OL].http://www.keinsci.com/molclus.html.research/.
[12]FRISCH M J,HEAD-GORDON M,Trucks G W,et al.Gaussian 90[CP].Gaussian Inc,Pittsburgh,1990.
[13]MASON G.Radial distribution functions from small packings of spheres[J].Nature,1968,217:733-735.
[14]DAI J X,LI X F,ZHAO L F,et al.Enthalpies of mixing predicted using molecular dynamics simulations and OPLS force field[J].Fluid Phase Equilibria,2010,289(2):156-165.
[15]XU W,YANG J.Computer simulations on aggregation of acetic acid in the gas phase,liquid phase,and supercritical carbon dioxide[J].The Journal of Physical Chemistry A,2010,114(16):5377-5388.
[16]SAIZ L,PADRO J A,GUARDIA E.Structure of liquid ethylene glycol:a molecular dynamics simulation study with different force fields[J].The Journal of Chemical Physics,2001,114(7):3187-3199.
GEMC simulation and microstructure analysis of vapor-liquid equilibrium for diol mixtures
LI Hong1,2,LI Dongyang1,2,LI Xingang1,2,3,GAO Xin1,2
(1School of Chemical Engineering and Technology,Tianjin University,Tianjin 300072,China;2National Engineering Research Centre of Distillation Technology,Tianjin 300072,China;3Collaborative Innovation Center of Chemical Science and Engineering(Tianjin),Tianjin 300072,China)
Gibbs ensemble Monte Carlo simulation(GEMC)method is used to investigate the vapor-liquid equilibria and microstructure of ethylene glycol(EG)/1,2-butanediol(1,2-BDO),EG/1,3-but-anediol(1,3-BDO)and EG/1,4-butanediol(1,4-BDO)mixtures.The simulation approach and OPLS-AA force field are proved to be adaptable to calculate the vapor-liquid equilibria by comparing the simulating results with experimental data or results calculated from Wilson EOS.Analyses of radial distribution functions for the mixtures above show that there is strong hydrogen-bonding interaction that is unrelated to the site of hydroxyl and Van der Waals interaction occurred in different regions among diol-mixtures.Statistics of the number of O-O atom pair and analyses of the hydrogen-bonding association structure reveal that with increasing EG in the mixture,the microstructure of the saturated liquid phase changes from the state consisting of many big BDO-BDO and EG-BDO hydrogen-bonding association structure(>6)to that composed of many small EG-EG and EGBDOhydrogen-bonding association structure(2~4)with decreased proportion of associated molecules.Molclus and Gaussian09 are utilized to search for the stable structure of the EG,1,2-BDO small hydrogen-bonding configuration.Then,one possible reason is proposed to explain why the proportion of diols association structure changes in some law above.That is,1,2-BDO-1,2-BDO association structure behaves more stable than others,and,as the number of associated molecules grows,the associated structure changes to be more stable,while the difference between the stability of big EG-EG association structures and smaller EG-1,2-BDO association structures decreases.
Gibbs ensemble Monte Carlo simulation;ethylene glycol/1,2-butanediol(EG/1,2-BDO);vapor-liquid equilibrium;microstructure
O 641.3;O 642.4+2
A
1000-6613(2016)09-2670-08
10.16085/j.issn.1000-6613.2016.09.006
2016-01-04;修改稿日期:2016-01-28。
国家自然科学基金项目(21306128)。
李洪(1980—),女,特聘研究员。E-mail lihong.tju@163.com。联系人:高鑫,副教授。E-mail gaoxin@tju.edu.cn。
猜你喜欢
中国音乐学(2022年1期)2022-05-05
科学家(2022年3期)2022-04-11
冰雪运动(2021年2期)2021-08-14
广州化工(2020年5期)2020-04-01
物理学报(2018年10期)2018-06-14
中国盐业(2018年23期)2018-03-30
中国塑料(2016年1期)2016-05-17
中国塑料(2016年11期)2016-04-16
中国塑料(2016年5期)2016-04-16
中国塑料(2015年5期)2015-10-14
