
烟叶表面微生物类群两种检测方法的比较研究
2016-11-29 04:06刘玉配李媛媛
华东师范大学学报(自然科学版) 2016年3期
龚 俊, 刘玉配, 李媛媛,2
(1.华东师范大学生态与环境科学学院浙江天童国家森林生态系统野外观测研究站,上海 200241; 2.上海烟草集团有限责任公司,上海 200082)
烟叶表面微生物类群两种检测方法的比较研究
龚 俊1, 刘玉配1, 李媛媛1,2
(1.华东师范大学生态与环境科学学院浙江天童国家森林生态系统野外观测研究站,上海 200241; 2.上海烟草集团有限责任公司,上海 200082)
为研究克隆文库法和高通量测序两种方法对烟叶表面微生物类群的检测效果,确定检测烟叶表面微生物多样性的最优方法,检测了基于细菌16S rDNA和真菌ITS区域的烤烟表面微生物类群的多样性.结果显示,高通量测序得到的序列数量比克隆文库法多,两种方法检测到细菌的OTU数目相近,但高通量检测的真菌OTU数目较多;稀疏曲线显示高通量测序的数据饱和度更高;预计的群落多样性(Chao1指数和Ace指数)克隆文库法均高于高通量测序法,实际的群落多样性(Simpson指数和Shannon指数)表明克隆文库法检测的细菌多样性略高,而高通量测序检测的真菌多样性略高;在检测到的细菌和真菌种类上,高通量测序略多于克隆文库法,两种方法检测的细菌优势类群基本一致,为假单胞菌属、不动杆菌属和鞘氨醇单胞菌属,但真菌类群相差较大,且高通量测序检测到大量新的真菌序列.总体上,高通量测序方法具有通量大、产出数据多的优势,可更全面、更准确地反映烟叶表面微生物群落结构.
克隆文库; 高通量测序; 细菌; 真菌; 烟叶
0 引 言
烟叶陈化是烟草加工过程中改善烟叶品质的一个重要环节,其实质是在一定环境条件下,烟叶内部理化特性发生缓慢而充分的变化.目前认为烟叶发酵机理主要包括酶作用、微生物作用和化学作用三个方面[1].其中烟叶表面微生物不仅自身的生命活动作用于烟叶的整个陈化过程,还通过代谢产生的生物酶参与烟叶的生理生化反应,促进烟叶生物大分子化合物的转化及致香成分的产生和积累,同时降低烟草中的有害物质,使烟叶品质得到提高[2].因此,对烟叶表面微生物类群的研究可客观深入地了解烟叶化学成分的变化规律.
早期对烟叶发酵过程中微生物群落多样性的研究主要通过平板分离并培养可培养微生物来确定优势菌群[3-4].但存在不同微生物的增殖差异问题,并且99%的微生物不能在实验室条件下培养出来[5-6],因而难以客观反映烟叶表面微生物类群状况.随着分子生物学快速发展,利用变性梯度凝胶电泳(Denaturing Gradient Gel Electrophoresis,DGGE)和限制性内切酶片段长度多态性(Restriction Fragment Length Polymorphism,RFLP)等分子技术鉴定烟叶发酵过程中微生物种类的研究也越来越多[6-7].然而,这些方法通过观察挑选电泳条带进行测序分析,局限于条带的浓度,得到的结果不完全[8],因此也不能充分反映烟叶表面的微生物种类.
克隆文库法通过构建文库并选择一定数量的克隆进行测序,一定程度上弥补了无法鉴定不可培养微生物的缺陷,近年来已较多地用于烟叶表面微生物类群鉴定[9-11].随着二代测序技术的发展,一种新的研究环境微生物群落多样性的方法——宏基因组学应运而生,基于高通量测序对环境微生物群落多样性进行研究,可直接对从环境中获得基因组样品进行测序分析,具有通量大、产出数据多、更为高效的优势[12],但目前鲜见应用于烟叶表面微生物的检测.本研究应用克隆文库法和高通量测序两种方法检测烤烟烟叶表面微生物类群多样性,比较两种方法检测结果的差异,筛选优势环境微生物鉴定方法,为后续环境微生物区系及动态变化研究提供基础.
1 材料与方法
1.1 实验材料
2013年10月采集产地为福建、等级为C/挑的烤烟烟叶样品,装入无菌自封袋中于-72℃保存,以备提取烟叶表面微生物DNA.
1.2 微生物收集
参考Zhao等[6]和Su等[9]的方法收集烟叶表面微生物,具体步骤为:称取40 g烟叶样品剪碎,置于装有500 mL灭菌的0.1 mol/L磷酸盐缓冲液(pH 7.0)中,于27℃、210 r/ min的摇床上振荡30 min,然后用单层纱布过滤,可得悬浮在磷酸缓冲液内的叶表面大部分微生物.滤液用大容量离心机10 000×g离心30 min,收集沉淀即为烟叶表面微生物.
1.3 微生物总DNA提取
采用细菌基因组DNA提取试剂盒(离心柱型)(TIANGEN公司)进行烟叶表面微生物总DNA的提取,即可得到烟叶表面细菌和真菌的总DNA.
1.4 克隆文库法鉴定微生物类群
1.4.1 PCR扩增
细菌16S rDNA序列扩增采用引物799F(5′-GGTAGTCCACGCCGTAAACGATG-3′)和1492R(5′-GGTTACCTTGTTACGACTT-3′),扩增片段长度约715 bp;真菌的转录间隔区ITS序列的扩增采用引物ITS4(5′-TCCTCCGCTTATTGATATGC-3′)和ITS5(5′-GGAAGTAAAAGTCGTAACAAGG-3′),扩增片段长度约500 bp.PCR反应体系如下, 150 ng模板DNA,1.25U Ex Taq聚合酶(Takara公司),1×Ex Taq Buffer(Mg2+plus), 200μmol/L dNTP Mixture,0.2μmol/L引物,补加灭菌ddH2O至50μL.PCR反应程序如下,94℃5 min;(94℃1 min,Tm 45 s,72℃1 min)30个循环;72℃8 min.其中扩增16S r DNA序列的退火温度为53℃,ITS序列为56℃.
1.4.2 克隆文库构建和测序
PCR产物采用多功能DNA纯化回收试剂盒(离心柱型)(北京百泰克生物技术有限公司)纯化回收.将PCR纯化产物与p MD19-TVector(Takara公司)连接后转入TOP10感受态细胞中(TIANGEN公司),在氨节平板上37℃培养.挑选白色菌落用M13F和M13R载体引物扩增验证阳性克隆后,在上海生工生物工程有限公司进行双向测序.
1.5 高通量测序分析微生物类群
高通量测序在上海美吉生物工程有限公司进行.细菌16S rDNA和真菌ITS测序均在Roche 454 GS FLX+测序平台上进行.细菌和真菌测序区域均与克隆文库法相同,细菌测序引物是799F和1492R,真菌测序引物是ITS4和ITS5.
1.6 数据处理
克隆文库法测序得到的序列去载体、拼接、比对,采用B2C2[13]检测并去除嵌合体序列后得到用于后续分析使用的优化序列,采用mothur软件[14]对优化序列进行操作分类单元(operational taxonomic units,OTU)聚类,以97%相似性为阂值,得到OTU代表序列.高通量测序得到的序列用Qiime v1.17[15]去杂过滤得到有效序列后用uchime-ref方法[16]去除嵌合体序列后得到优化序列,在Usearch v7.1中调用uparse方法[17]对优化序列按相似度97%进行聚类,得到OTU代表序列.计算两种方法测序数据的文库覆盖率(Coverage),文库覆盖率是计算测序深度的指标[23],值越高表示样品中序列被测出来的概率越大,计算公式为:

其中,n1为只含有一条序列的OTU数,N为样品中的优化序列的数目.
对97%相似水平的OTU代表序列采用RDP Classifier方法[18-19]进行分类学分析,置信度阂值为95%.用mothur软件对97%相似度的OTU做稀有度分析,利用SigmaPlot v12.5制作稀疏曲线图.用mothur软件计算4种多样性指数:Chao1指数、Ace指数、Simpson指数和Shannon指数.Chao1和Ace指数是利用现有结果对整个群落的OTU多样性进行预测的指标,其越大代表群落多样性越高;Simpson和Shannon指数是对现有结果进行OTU多样性分析的指标,Simpson值越大说明群落多样性越低,Shannon指数值越大说明群落多样性越高.
Chao1指数[20]表示样品中所含估计OTU的数目,计算公式为:

其中,SChao1为估计的OTU数,Sobs为实际检测到的OTU数,n1为含有一条序列的OTU数, n2为含有2条序列的OTU数.
Ace指数[21-22]是用来估计群落中OTU数目的指数,计算公式为:
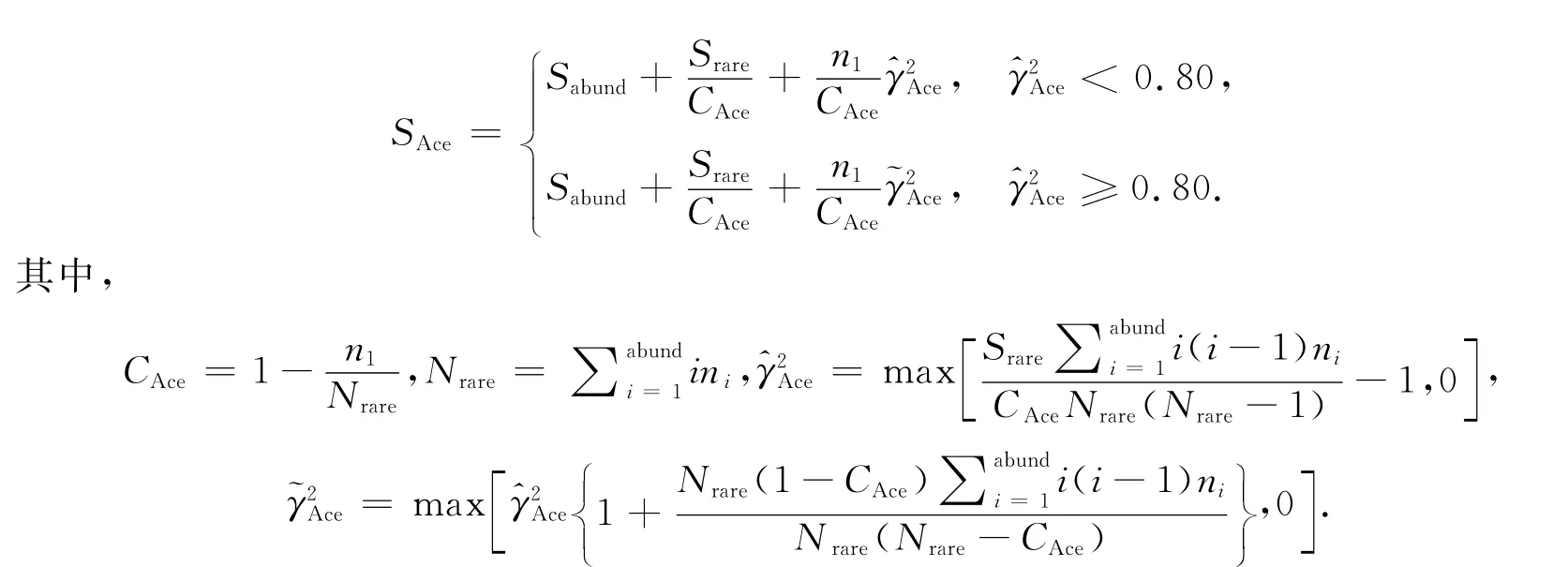
ni为含有i条序列的OTU数目;Srare为含有或少于“abund”条序列的OTU数目;Sabund为多于“abund”条序列的OTU数目;abund为划分优势OTU的序列数量阂值,默认10.
2 结 果
2.1 序列优化与OTU聚类
细菌16S rDNA:克隆文库法获得有效序列152条,优化后序列共141条,以97%序列相似性为阂值,可聚类为79个OTUs;而高通量测序获得有效序列高达10 017条,优化后得到4 861条,可聚类为76个OTUs;两种测序方法得到的序列应用率分别为92.76%和48.53%(见表1).
真菌ITS:克隆文库法获得有效序列88条,优化后共86条序列,以97%序列相似性为阂值,可聚类为18个OTUs;而高通量测序获得ITS有效序列5 742条,优化后得到4 754条,优化序列可分为37个OTUs.两种测序方法得到的序列应用率分别为97.73%和82.78%(见表1).
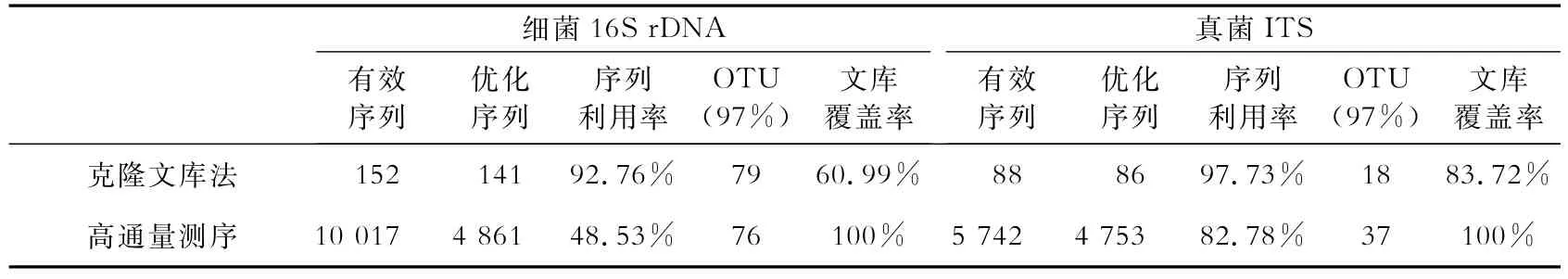
表1 序列优化及OTU聚类结果Tab.1 Results of trimmed sequences and OTU analysis
高通量测序法测得的细菌和真菌的覆盖率都为100%.克隆文库法细菌测序克隆数为163,其中测出序列152条,优化序列的文库覆盖率为60.99%;真菌测序克隆数为91,其中测出序列88条,优化序列的文库覆盖率为83.72%.
2.2 稀疏曲线
运用两种分析方法得到的细菌和真菌的稀疏曲线如图1所示.高通量测序得到的不管是细菌还是真菌的稀疏曲线都呈现逐渐平缓的趋势,说明数据饱和度较好;而克隆文库法得到的细菌和真菌的稀疏曲线都呈陡峭的上升趋势,表明数据饱和度不够,但序列间差异大,鉴别OTUs的比例高.
2.3 多样性分析
两种测序方法得到的多样性指数见表2.克隆文库法得到的细菌的Chao1指数、Ace指数和Shannon指数都较大,Simpson指数较小,说明克隆文库法得到的细菌群落预计的和实际的多样性都较高.克隆文库法得到的真菌的Chao1指数和Ace指数都较大,说明克隆文库法得到真菌群落预计的多样性较大,而Simpson指数和Shannon指数显示高通量测序得到的真菌实际多样性更高.

表2 细菌和真菌的多样性指数Tab.2 Diversity indices in bacterial and fungal communities
2.4 群落分类情况
将测序得到的序列归类到各分类水平后,统计结果如表3所示.已知和未知种类总体上来看,克隆文库法检测到细菌共有3门6纲11目20科28属,真菌共有3门6纲7目10科12属.高通量测序共检测到细菌有5门7纲16目30科44属,真菌共有3门8纲13目15科19属.从两种方法检测的比值来看,高通量测序检测到的细菌和真菌种类都高于克隆文库法,并且一般来说分类水平越低,检测到的种类越多.

表3 两种测序方法检测到的细菌和真菌分类情况Tab.3 Taxon of bacteria and fungi detected by two different methods
2.5 细菌群落结构与优势菌群
两种测序方法得到的细菌群落在门分类上的组成见图2.克隆文库法得到的细菌为3个门,分别为变形菌门(Proteobacteria)、厚壁菌门(Firmicutes)和放线菌门(Actinobacteria),还有部分序列不能被分类的归为Unclassified;高通量测序得到的细菌可分为5个门,除包括克隆文库法得到的3个门外还有蓝藻门(Cyanobacteria)和拟杆菌门(Bacteroidetes),以及不能分类的Unclassified,高通量测序鉴别的细菌门类多于克隆文库法.两种测序方法检测细菌门类最多的都是变形菌门,分别占90.78%和81.32%,得到的优势菌门相同.克隆文库法有部分细菌门如蓝藻门和拟杆菌门没有检测到.
两种方法得到的细菌群落在属分类上的结构见图3.克隆文库法检测到的已知分类的属为14个,高通量测序为36个,其中有11个属共同检测到.克隆文库法检测到类群最多的3个属分别是假单胞菌属(Pseudomonas)、不动杆菌属(Acinetobacter)和鞘氨醇单胞菌属(Sphingomonas),分别占9.22%、9.22%和6.38%;高通量测序检测类群最多的属分别是假单胞菌属、甲基杆菌属(Methylobacterium)、不动杆菌属和鞘氨醇单胞菌属,分别占11.24%、7.66%、6.29%和6.00%.在属的分类水平上,高通量测序可以检测到的细菌属种类更多,两种测序方法得到的优势菌属基本一致.
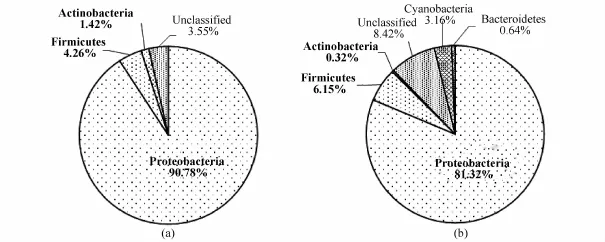
图2 细菌群落在门分类水平的结构Fig.2 Bacterial community structure on phylum level

图3 细菌群落在属分类水平的结构Fig.3 Bacterial community structure on genus level
2.6 真菌群落结构与优势菌群
两种测序方法得到的真菌群落在门分类上的组成见图4.真菌中担子菌门(Basidiomycota)、子囊菌门(Ascomycota)和接合菌门(Zygomycota)3个门在两种方法中都鉴定出来,都有部分序列不能被分类的归为Unclassified.其中担子菌门种类最多,达60%以上,其次是子囊菌门,最少的是接合菌门.
两种方法鉴定的真菌群落在属分类上的组成见图5.克隆文库法鉴定了10个已知分类的真菌属,高通量测序鉴定了11个已知分类真菌属,但是两种方法鉴定为相同的属只有1个,为曲霉属(Aspergillus).克隆文库法获得属的比例都1%,其中比例最高的3个属分别是红酵母属(Rhodotorula)、假丝酵母属(Candida)和根霉属(Rhizopus),分别占62.79%、19.77%和3.49%;高通量测序获得的序列大部分都未能分到现有属中(88.27%),比例1%的只有Meyerozyma属、散囊菌属(Eurotium)和翘抱霉属(Emericella)3个属,分别占5.37%、2.42%和2.13%.在属分类水平上,两种方法鉴定的真菌属种类相差很大.

图4 真菌群落在门分类水平的结构Fig.4 Fungal community structure on phylum level

图5 真菌群落在不同属分类水平的结构Fig.5 Fungal community structure on genus level
3 讨 论
烟叶发酵过程微生物种类繁多,也起到了不同的作用.本研究采用克隆文库法和高通量测序法对烟叶表面微生物种类进行检测,在序列数量和OTU种类上两种方法差异都较大.克隆文库法得到的序列数量与克隆的生长和测序数目有关,获得几百条序列已属大量,而高通量测序一次反应则至少能获得上千条序列,表明高通量测序具有通量大、产出数据多的优势[12].但细菌OTU划分结果显示两种方法获得的种类数量相差不多,分别为79(克隆文库法)和76(高通量测序)个,真菌OTU划分结果显示高通量测序获得的OTU种类(37个)多于克隆文库法(18个).本研究中高通量的OTU的建立采用较新的UPARSE方法,该方法比其他如QIIME和mothur等聚类出OTU的数量少,但在与已知种类的序列匹配、误差率和嵌合子(chimeric)出现的概率等方面都有优势[17].
从两种测序方法得到的文库覆盖率可以看出高通量测序得到的文库覆盖率更高,从稀疏曲线中也可以看出高通量测序得到的数据饱和度高,而克隆文库法鉴定的序列差异大,远未达到饱和,这一结果与UPARSE方法分析高通量数据时统计方法有关.UPARSE进行高通量数据统计时,对测出的单拷贝序列(只出现1次的单独1条序列,singleton)进行了优化去除[17],大大降低了序列的差异性,使得计算的文库覆盖率为100%,在稀疏曲线中也呈现饱和,增加了结果的保守性,而克隆文库法由于单拷贝序列的大量出现导致文库覆盖率降低,稀疏曲线无限增加[24].这也反映在预计的群落多样性指数(Chao1指数和Ace指数)上,克隆文库法鉴定的细菌和真菌的预计多样性均高于高通量测序法,显示了保守性.而实际的群落多样性(Simpson指数和Shannon指数)克隆文库法得到的细菌多样性略高,真菌的多样性高通量测序的略高(见表2).类似Chao1指数这类多样性指标较多地依赖于单拷贝序列的数目,在高通量测序数据的分析中受到质疑[25].
从微生物组成情况可以看出,高通量测序检测到的细菌和真菌的已知种类均多于克隆文库法,且分类水平越低,检测到的微生物种类越多,在门分类水平上两种测序方法得到的细菌种类差异最小,在科、属水平上差异最大(见表3).两种方法检测的细菌优势门相同,都为变形菌门(都占80%以上),其次是厚壁菌门,与Su等用克隆文库法[9]、Huang等用RFLP法[7]检测烟叶表面细菌的优势门一致.在属分类水平上,本研究两种方法得到的优势细菌类群基本一致,为假单胞菌属、不动杆菌属和鞘氨醇单胞菌属,高通量测序还有甲基杆菌属也属优势类群.Su等[9]用克隆文库法检测到津巴布韦烟叶表面细菌可分为16个属,其中有8个属与本文检测结果一致,优势菌属为泛菌属和假单胞菌属,与本文检测也基本一致.Huang等[7]采用RFLP法检测到未陈化烤烟K326表面细菌可分为50种,优势菌属为芽抱杆菌属和假单胞菌属.段焰青等[26]采用平板分离得到烟叶样品中的细菌包括芽抱杆菌属、类芽抱杆菌属、肠杆菌属和泛菌属与本文检测结果一致,另有柠檬酸杆菌属(Citrobacter)和欧文氏菌属(Erwinia)未在本研究中发现.
本研究两种方法检测的真菌优势门也一致,都为担子菌门和子囊菌门.但属的差异甚大,两种方法只有一个共同属,优势类群也不同.本文高通量测序得到的真菌属与段焰青等[26]分离得到的曲霉菌属和镰刀菌属一致,克隆文库法得到的根霉属在段焰青等[26]研究中也分离得到了,只有木霉属(Trichoderma)与本文结果有差异.另外,在高通量测序方法中有大量的序列没有归并到已知分类的属中(unclassified,88.27%),表明检测到大量在数据库中还未出现的真菌序列,这种情况在Edgar对真菌的检测中也曾出现[17].
两种测序方法得到的微生物多样性存在一定差异,导致这种差异的原因主要有:①导入目标DNA片段的大肠杆菌在培养基上成功生长存在随机性,导致某些微生物种类没有检测到;②克隆文库法存在文库构建和测序数量的限制导致获得序列数不足,因而不如高通量测序法更能充分反映环境中微生物组成;③用于高通量的焦磷酸测序本身存在的固有测序误差也会导致一些稀有微生物种类的出现[12],尤其是由于单核昔酸重复(均聚体)易出现而导致ITS序列在454测序中容易出错[27].总体上,高通量测序具有通量大、产出数据多的优势,因而可以更全面、更准确地反映烟叶表面微生物种类和结构的特点,且随着高通量测序方法日趋成熟、测序成本不断降低,该方法将有望成为今后研究烤烟微生物多样性的一种常用技术手段.
[1] 于建军,李琳,庞天河,等.烟叶发酵研究进展[J].河南农业大学学报,2006,40(1):108-112.
[2] 刘萍,张广民,郑小嘎,等.烟叶表面微生物及其应用[J].微生物学通报,2003,30(6):105-110.
[3] 韩锦峰,朱大恒,刘卫群,等.陈化发酵期间烤烟叶面微生物活性及其应用研究[J].中国烟草科学,1997(4):15-16.
[4] 邱立友,赵铭钦,岳雪梅,等.自然发酵烤烟叶面微生物区系的分离鉴定[J].烟草科技,2000(3):14-17.
[5] AMANN R I,LUDWIG W,SCHLEIFER K H.Phylogenetic identification and in situ detection of individual microbial cells without cultivation[J].Microbiological Reviews,1995,59(1):143-169.
[6] ZHAO M,WANG B,LI F,et al.Analysis of bacterial communities on aging flue-cured tobacco leaves by 16S r DNA PCR-DGGE technology[J].Applied Microbiology and Biotechnology,2007,73(6):1435-1440.
[7] HUANG J,YANG J,DUAN Y,et al.Bacterial diversities on unaged and aging flue-cured tobacco leaves estimated by 16S r RNA sequence analysis[J].Applied Microbiology and Biotechnology,2010,88(2):553-562.
[8] 夏围围,贾仲君.高通量测序和DGGE分析土壤微生物群落的技术评价[J].微生物学报,2014(12):1489-1499.
[9] SU C,GU W,ZHE W,et al.Diversity and phylogeny of bacteria on Zimbabwe tobacco leaves estimated by 16S r RNA sequence analysis[J].Applied Microbiology and Biotechnology,2011,92(5):1033-1044.
[10] 伍雪莹,梁书利,韩双艳,等.不同陈化期烤烟叶表细菌的多样性及发育分析[J].广东农业科学,2014(18):28-33.
[11] 陈竹亭,焉婷婷,汤朝起,等.应用16S rDNA克隆文库技术分析陈化烟叶细菌多样性[J].中国烟草学报,2012 (4):77-82.
[12] SIMON C,DANIEL R.Metagenomic analyses:Past and future trends[J].Applied and Environmental Microbiology,2011,77(4):1153-1161.
[13] GONTCHAROVA V,YOUN E,WOLCOTT R D,et al.Black box chimera check(B2C2):A windows-based software for batch depletion of chimeras from bacterial 16S r RNA gene datasets[J].The Open Microbiology Journal,2010(4):47-52.
[14] SCHLOSS P D,WESTCOTT S L,RYABIN T,et al.Introducing mothur:Open-source,platform-independent, community-supported software for describing and comparing microbial communities[J].Applied and Environmental Microbiology,2009,75(23):7537-7541.
[15] CAPORASO J G,KUCZYNSKI J,STOMBAUGH J,et al.QIIME allows analysis of high-throughput community sequencing data[J].Nature Methods,2010,7(5):335-336.
[16] EDGAR R C,HAAS B J,CLEMENTE J C,et al.UCHIME improves sensitivity and speed of chimera detection [J].Bioinformatics,2011,27(16):2194-2200.
[17] EDGAR R C.UPARSE:Highly accurate OTU sequences from microbial amplicon reads[J].Nature Methods, 2013,10(10):996-998.
[18] COLE J R,WANG Q,FISH J A,et al.Ribosomal database project:Data and tools for high throughput r RNA analysis[J].Nucleic Acids Research,2013,42(D1):D633-D642.
[19] WANG Q,GARRITY G M,TIEDJE J M,et al.Naive bayesian classifier for rapid assignment of r RNA sequences into the new bacterial taxonomy[J].Applied and Environmental Microbiology,2007,73(16):5261-5267.[20]CHAO A.Nonparametric estimation of the number of classes in a population[J].Scandinavian Journal of Statistics,1984:265-270.
[21]CHAO A,HUANG W,CHEN Y C,et al.Estimating the number of shared species in two communities[J]. Statistica Sinica,2000,10(1):227-246.
[22]CHAZDON R L,COLWELL R K,DENSLOW J S,et al.Statistical methods for estimating species richness of woody regeneration in primary and secondary rain forests of NE Costa Rica[M]//Forest Biodiversity Research, Monitoring and Modeling:Conceptual Background and Old World Case Studies.Paris:Parthenon Publishing, 1998:285-309.
[23]GOOD I J.The population frequencies of species and the estimation of population parameters[J].Biometrika, 1953,40:237-264.
[24]QUINCE C,LANZEN A,CUITIS TP,et al.Accurate determination of microbial diversity from 454 pyrosequencing data[J].Nature Method s,2009(6):639-641.
[25]DICKIE I A.Insidious effects of sequencing errors on perceived diversity in molecular surveys[J].New Phytologist,2010,188:916-918.
[26]段焰青,者为,李祖红,等.醇化烟叶表面微生物的多样性和系统发育分析[C]//中国烟草学会工业专业委员会工艺学组2008年学术研讨会论文集.吉林延吉:中国烟草学会,2008:85-89.
[27]LINDAHL B D,NILSSON R H,TEDERSOO L,et al.Fungal community analysis by high throughput sequencing of amplified markers:A user,s guide[J].New Phytologist,2013,199:288-299.
(责任编辑:张晶)
Comparative analysis of microbial communities on tobacco leaves between clone library and high-throughput sequencing
GONG Jun1, LIU Yu-pei1, LI Yuan-yuan1,2
(1.Tiantong National Eield Observation Station for Eorest Ecosystem,School of Ecological and Environmental Sciences,East China Normal University,Shanghai 200241,China; 2.Shanghai Tobacco Group Corporation,Shanghai 200082,China)
In order to compare the microbial communities on tobacco leaves between clone library method and high-throughput sequencing method,we analyzed the diversity of microbial communities based on bacterial 16S rDNA and fungal ITS on tobacco leaves collected from Fujian province in China.The results showed that more numbers of bacterial and fungal sequences were detected by high-throughput sequencing than clone library.The number of operational taxonomic units(OTUs)clustered from high-throughput sequencing method was almost the same as thatfrom clone library method in bacterial communities,while that was higher from high-throughput sequencing in fungal communities.The rarefaction curves drawn from high-throughput sequencing method tended to approach the saturation plateau.The expected community diversity indices (Chao1 and Ace)were higher in clone library method,whereas the observed community diversity indices(Simpson and Shannon)suggested that higher bacterial diversities were detected through clone library method and higher fungal diversities were detected through high-throughput sequencing method.There were more kinds of bacteria and fungi genera identified through high-throughput sequencing method.The dominant genera of bacteria were similar detected from both of the methods,including Pseudomonas,Acinetobacter and Sphingonomonas.However,the fungal genera detected through these two methods were significantly different.Overall,novel high-throughput sequencing methods outperform clone library approaches in terms of resolution and magnitude.They enable identification and relative quantification of community members of tobacco leaves and offer new insights into environmental microbiology.
clone library; high-throughput sequencing; bacteria; fungi; tobacco leaves
Q33
A
10.3969/j.issn.1000-5641.2016.03.011
1000-5641(2016)03-0092-10
2015-04
烟草行业卷烟烟气重点实验室开放基金(SZBCW2013-00596)
龚 俊,女,硕士研究生,研究方向为分子生态学.E-mail:gongjun.2009163.com.
李媛媛,女,博士,副教授,研究方向为分子生态学.E-mail:yylides.ecnu.edu.cn.
猜你喜欢
猪业科学(2021年3期)2021-05-21
透析与人工器官(2020年1期)2020-11-16
幽默大师(2020年10期)2020-11-10
中华诗词(2019年1期)2019-11-14
铁道通信信号(2019年8期)2019-10-10
活力(2019年15期)2019-09-25
猪业科学(2018年4期)2018-05-19
现代园艺(2017年23期)2018-01-18
中国发展观察(2017年8期)2017-04-26
中国当代医药(2015年33期)2015-03-01
