
基于RNA-seq的黄蜻头部转录组测序与分析
2016-12-09 10:06葛怡情曹玲珍
四川动物 2016年6期
葛怡情, 曹玲珍
(江西师范大学生命科学学院,鄱阳湖湿地与流域研究教育部重点实验室,南昌330000)
基于RNA-seq的黄蜻头部转录组测序与分析
葛怡情, 曹玲珍*
(江西师范大学生命科学学院,鄱阳湖湿地与流域研究教育部重点实验室,南昌330000)
利用RNA-seq技术对黄蜻Pantalaflavescens(Fabricius)头部细胞的全部转录本进行测序、功能分析。提取黄蜻头部总RNA,反转录得到cDNA,在Illumina平台上进行测序,经拼接、组装、聚类后获得全部unigenes,并将所得的unigenes与数据库比对,对其进行功能注释和分类。总共得到47 039 040条碱基序列,包含了7 102 895 040个碱基序列信息,碱基G和C的数量总和占总碱基数量的44.05%。将测序结果进行组装,组装所得到的unigenes为34 406 502条,对所得到的unigenes进行不同的数据库注释,共有44 499条unigenes被注释到数据库,其中有大量unigenes与代谢过程、结合活性、催化活性和细胞进程有关。与COG数据库比对,根据功能可以分为26类,其中信号转导机制最为丰富。与KEGG数据库比对,线粒体基因在所测样本中最为丰富,与KEGG ORTHOLOGY注释后,代谢活动基因最为丰富。这些与线粒体和代谢有关的基因与我们研究中与黄蜻迁飞基因相关,为后续研究奠定了基础。
黄蜻;转录组;测序;基因分析;功能注释
蜻蜓目Odonata是一类比较古老的昆虫,是天敌昆虫和药用、食用资源昆虫,也是水体污染程度的指示昆虫之一(张大治,杨贵军,2006)。黄蜻Pantalaavescens(Fabricius)隶属于蜻蜓目蜻科Libellulidae黄蜻属Pantala,在世界各地的热带和温带都有发现。黄蜻有一个极短的稚虫期(34~43 d;Suhlingetal.,2004),可以在热带辐合带降雨产生的临时水池中繁殖。成虫被认为可以随着热带辐合带气流进行广泛的迁飞(Hobsonetal.,2012)。在一些分布区内,黄蜻属于季节性分布,其在迁飞或者扩散中常聚成大群(Fengetal.,2006)。蜻蜓目主要取食蚊、蝇、叶蝉、小型蛾子、蝴蝶、虻等各种小型昆虫。一只普通蜻蜓1 h可吃掉40只苍蝇或840只蚊子。蜻蜓稚虫生活在水中,捕食蚊子幼虫等水生动物,其对迁飞害虫的生物防治起到了一定的作用,有利于农业、园林的害虫防治。以蜻蜓为材料认识迁飞在蜻蜓生活史中的作用,还能有效地对害虫进行控制和预防。
迄今为止,国外关于蜻蜓目昆虫多样性研究主要从外部形态学标记和分子标记展开。关于迁飞技术的研究发展方面,人们常利用昆虫的趋光性,用诱虫灯对迁飞昆虫进行诱捕(刘立春,1994)。20世纪30—40年代,英国昆虫学家借助风筝对白天飞行的昆虫进行了取样研究(Hardy & Milne,1938); Chapman等(2004)利用气艇在英国南部对迁飞昆虫群落进行了取样分析; 20世纪70年代,全国褐飞虱Nilaparvatalugens科研协作组在全国设置了40多个高山捕虫网(海拔>2 000 m),开展了褐飞虱和白背飞虱Sogatellafurcifera的研究(邓望喜,1981);近年来,通过用永久性标记在翅上写号码做标记,容易获得样品(van Noordwijk,1978;Watanabeetal.,2004)。Wikelski等(2006)在巴拿马的巴罗科罗拉多岛上,通过在碧伟蜓Anaxparthenopejulius腹部安装1个300 mg信号发射器来研究其南迁现象,但由于碧伟蜒的中途停留、迁飞的不确定性以及标记个体的死亡,该方法难度较大,效果不是很理想。微卫星DNA广泛分布于原核和真核生物中,具有重复性好、共显性遗传、遗传多态性高、操作简单快速及结果可靠等优点,使其在物种的遗传和进化中被广泛使用(Freelandetal.,2003;Matthews, 2007; Matthewsetal.,2007;Wellenreutheretal.,2011)。稳定的同位素分析被一致认为是研究脊椎动物和非脊椎动物种群迁飞的一种比较好的手段(Hobsonetal.,2010,2012)。“转录组”最先由Velculescu等(1997)提出,为细胞功能的诠释提供了更高的价值信息(吴琼等,2010),比传统基因挖掘更具优势(张楠等,2013)。本研究将Illumina Hiseq 4000高通量测序技术应用到黄蜻转录组研究,将测序得到的大量数据进行拼接与组装,结合生物信息学方法对所获得的unigenes进行基因功能注释、功能分类、代谢途径分析等,从功能基因组水平上研究黄蜻迁飞过程中重要基因的表达,使人们能够了解其在生态系统中的作用,有利于物种保护和生物多样性的形成,同时能够对一些害虫进行有效控制。
1 材料与方法
1.1 研究对象
2015年7月于江西省南昌市郊捕捉黄蜻20只。本研究中所采用的都是成熟个体;雄性个体居多(前期调查发现迁飞黄蜻的性比接近1∶1,雌雄个体对本研究影响不大)。取其头部,冷冻于液氮中。
1.2 提取总RNA
从黄蜻头部提取总RNA,利用Nanodrop 2000对所提RNA的浓度和纯度进行检测,琼脂糖凝胶电泳检测RNA完整性。
1)取50~100 mg黄蜻头部,于液氮预冷的研钵中,加入液氮研磨至粉末状后倒入离心管中,加入2 mL Trizol震荡30 s,静置30 s,加入1/5体积的氯仿,震荡30 s,静置1 min。12 000 r·min-1,4 ℃离心5 min。
2)小心地吸取上层液相,并避免接触到下层液相,加入1/2体积的异丙醇,震荡30 s,静置1 min,12 000 r·min-1,4 ℃离心5 min。
3)弃上清液,加入1 mL 75%乙醇,翻转洗涤沉淀RNA, 12 000 r·min-1,4 ℃离心5 min。
4)将沉淀的样品置于超净工作台上晾干,加入适量的DEPC H2O溶解(60 ℃促溶10 min)。
1.3 文库构建与测序分析
取黄蜻头部总RNA用微量紫外分光光度计检测其浓度和纯度,用带有Oligo(dT)的磁珠富集mRNA。加入破碎缓冲液,将获得的mRNA随机断裂成200 bp左右的小片段,在逆转录酶的作用下,利用随机引物,以mRNA为模板合成第一链cDNA,随后进行第二链合成,形成稳定的双链结构,再进行纯化回收、修复粘性末端、于cDNA的3’末端加上碱基‘A’并连接接头,然后进行片段大小选择,最后进行PCR扩增构建文库。制备好的文库用Illumina Hiseq 4000进行测序。原始测序结果去除制备文库时产生的接头序列、两端低质量序列和低度复杂序列。利用Trinity(http://trinityrnaseq.sourceforge.net)对样品数据进行组装,获得RNA-seq高质量测序数据后,将所有测序读段通过从头组装生成重叠群(contig)和单一序列(singleton)。
1.4 功能注释与序列分析
将拼接所得核苷酸序列,使用BlastX分别与NR、String、SwissProt、KEGG数据库进行比对获得相应的注释信息,共有44 499条unigenes被注释到数据库。
美国国立生物技术信息中心(NCBI,非冗余蛋白数据库)包含SwissProt和PRF(Protein Research Foundation)等蛋白质序列数据库。根据NCBI数据库的功能注释信息,得到unigene的GO条目,然后对所有的unigenes进行GO(Gene Ontology;http://www.geneo-ntology.org)功能分类统计与GO数据库比对,对应到GO的生物过程(biological process)、生物组分(cellular component)和分子功能(molecular function)。这3个大分支下面又分很多小层级(level),level级别数字越大,功能越细致。通过与NR数据库比对,查看本物种转录本序列与相近物种相似的程度,以及同源序列的功能信息。然后对unigenes分别进行蛋白质直系同源数据库(Cluster of Orthologous Groups,COG;http://www.ncbi.nlm.nih.gov/COG/)功能分类,与COG数据库比对可以进行功能注释、归类以及对相关蛋白进化分析。与东京基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG;http://www.genome.jp/kegg/)代谢途径分析,通过与KEGG数据库比对,获得转录本对应的KO编号,根据KO编号可以获得某转录本中一些基因的具体生物学通路,注释后,可根据它们参与的KEGG代谢通路进行归类。通过上述数据库对基因注释,找到与黄蜻迁飞运动过程相关基因的表达。
2 结果与分析
2.1 测序结果与数据组装
采用Illumina Hiseq 4000技术对黄蜻头部转录组进行测序,共获得45 908 046个片段,其中包含了6 694 354 464个核苷酸序列信息,碱基错误率为0.013 9%,G、C百分含量为44.05%,Q20为97.34%,Q30为91.64%。数据表明,转录组测序数据量和质量都较高。对所得的reads进行转录组组装,得到转录本47 188条,unigenes 37 868条,其中,转录本长度为1~600 bp的有25 701条,占总体的54.47%;长度为1 001~5 000 bp的有14 480条,占30.69%;长度>10 000 bp仅占0.17%;unigenes长度为1~600 bp的有23 457条,占总体的61.94%,长度为1 001~5 000 bp的有9 385条,占24.78%,长度>10 000 bp的仅占0.09%(表1)。
2.2 unigenes的功能注释结果
2.2.1 NR数据库注释结果 黄蜻的unigenes比对到最多的近缘物种有内华达古白蚁Zootermopsis
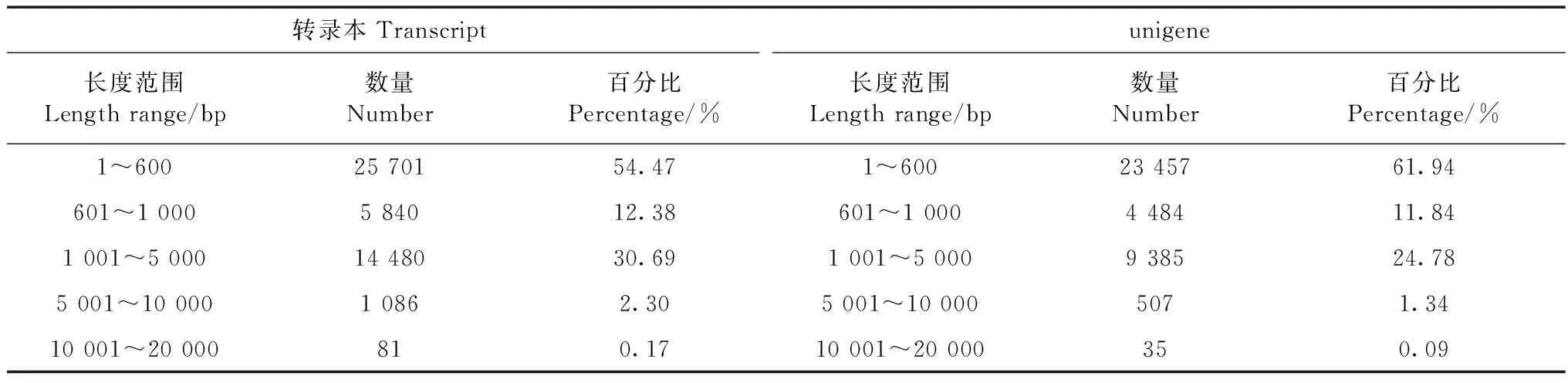
表1 黄蜻头部转录组转录本和unigene数据组装质量统计
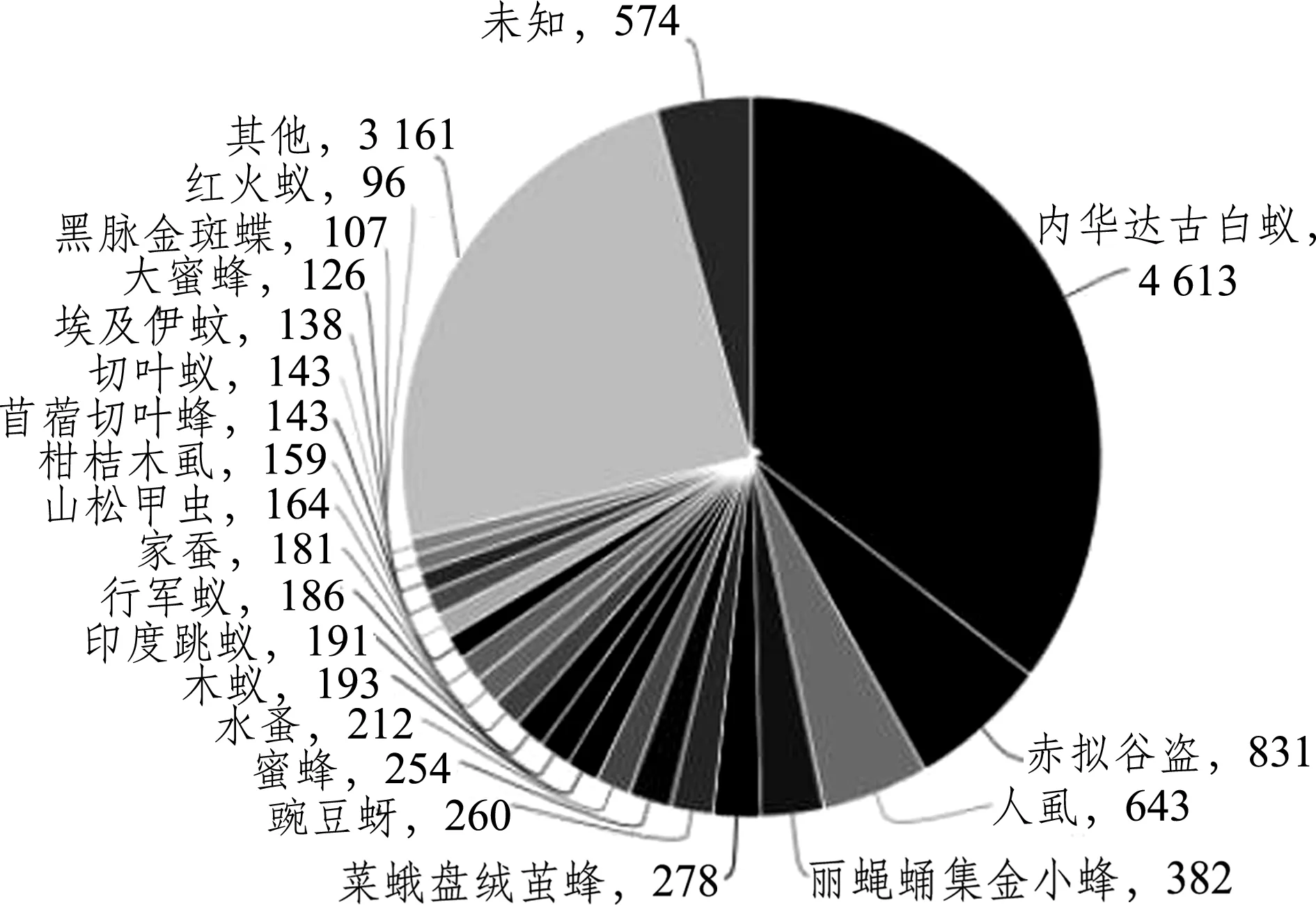
图1 物种分类
nevadensis(4 613),其次是赤拟谷盗Triboliumcastaneum(831)(图1)。在区间0内,基因数量最多(7 503),匹配结果也最高,而在(1e-10,1e-5]区间内,基因数量最少(919);相似度在80%~100%区间内的基因数量最多(6 281),而在20%~40%区间内的基因数量最少(35)(图2)。
2.2.2 GO注释结果统计 黄蜻的unigenes分为生物过程、生物组分和分子功能3个本体,二级分支共60个功能组,注释到生物过程的unigenes最多。在生物过程中,注释最多的是细胞的进程(cellular process)和代谢过程(metabolic process),而激素分泌(hormone secretion)、生物相(biological phase)、细胞杀伤(cell killing)等几乎没有。
生物过程二级分支中,与细胞的进程相关最多,有3 292条与其对应,与细胞组织生物合成(cellularcomponent organization or biogenesis)相关最少,有564条与其相对应;三级分支中,与有机物质代谢过程(organic substance metabolic process)相关最多,有2 518条与其相对应,与建立定位(establishment of localization)相关最少,有858条与其相对应;四级分支中,与大分子代谢过程(macromolecule metabolic process)相关最多,有1 764条与其相对应,而与有机物生物合成过程(organic substance biosynthetic process)相关最少,有1 021条与其相对应。
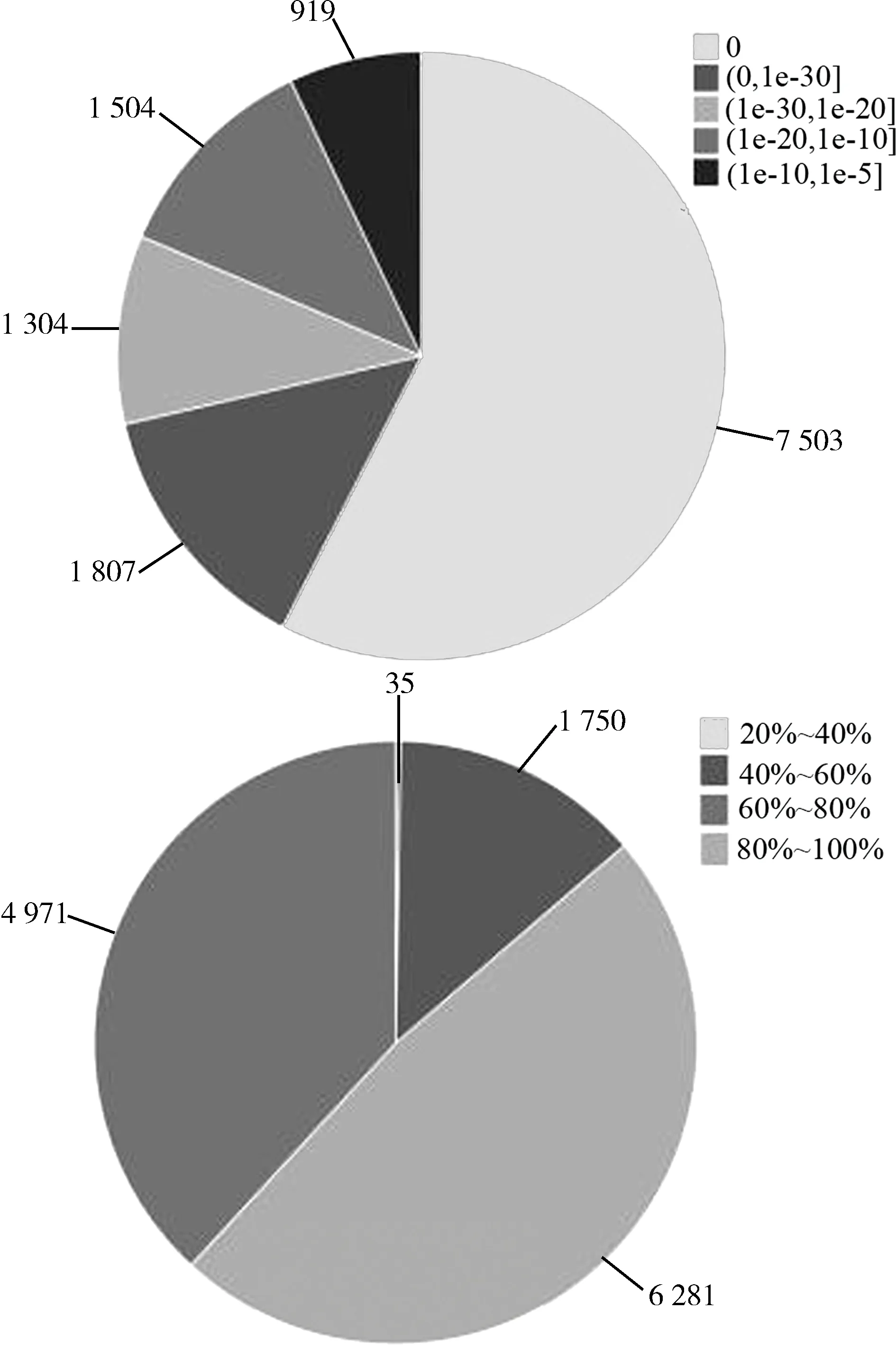
图2 比对结果e-value分布相似度分布
生物组分二级分支中,与细胞(cell)和细胞的部分(cell part)相关最多,有1 901条,胞外区域部分(extracellular region part)相关最少,有174条;三级分支中,与细胞的部分相关最多,有1 901条,与细胞器部分(organelle part)相关最少,有640条;四级分支中,与细胞内的(intracellular)相关最多,有1 716条,与细胞内的非膜结合细胞器(intracellular non-membrane-bounded organelle)相关最少,有616条。
分子功能二级分支中,与结合(binding)相关最多,有2 782条,与蛋白质结合转录因子活性(protein binding transenption factor activity)相关最少,有38条;三级分支中,与有机环状化合物结合(organic cyclic compound binding)和杂环化合物的结合(heterocyclic compound binding)相关最多,有1 741条,与跨膜转运蛋白活性(transmembrane transporter activity)相关最少,有329条;四级分支中,与核苷酸结合(nucleotide binding)和核苷磷酸结合(nucleotide phosphate binding)相关最多,有957条,与特定底物的跨膜转运活动(substrate-specific transmembrane transporter activity)相关最少,有286条(图3)。
2.2.3 ORF预测 利用Trinity软件提供的ORF流程对组装得到的转录本进行基因预测,得到大量的蛋白质与核苷酸序列,找出其在组装结果中的相对位置,并对转录本进行蛋白质家族预测,有26 561条unigenes预测到了蛋白质,有44 262条转录本预测到了蛋白质。
2.2.4 COG注释结果 黄蜻的unigenes根据其功能可分为26类,其中信号转导(signal transduction mechanisms)最丰富,其次为转录(transcription)、一般功能预测(general function prediction only)、翻译后修饰、蛋白质折叠和分子伴侣(posttranslational modification, protein turnover, chaperones)(图4)。
2.2.5 KEGG通路注释结果 根据序列比对分析,获得转录本参与的具体生物学通路,其中注释数量较多的有核糖体(ribosome)(222条)、癌症通路(pathways in cancer)(183条)、HTLV-I感染(HTLV-I infection)(179条)、舞蹈病(Huntinngton’s disease)(177条)、P13K-Akt信号通路(P13K-Akt signaling pathway)(176条)。根据它们在KEGG中的代谢通路进行分类,共有A(代谢)、B(遗传信息处理)、C(环境信息处理),D(细胞过程)和E(有机系统)五大类。在这五大类中代谢类最多,而代谢中又以全球概览图(global and overview maps)含量最高,共有1 067个(图5)。
3 讨论
RNA-seq技术结合了转录组测序建库的实验方法与数字基因表达谱(digital gene expression profiling,DCE)的信息分析手段,具有定量准、可重复性高、检测范围宽等特点(朱立强,李庆花,2016)。Illumina高通量测序的数据量大、速度快、成本低、效率高(贾新平等,2014)。黄蜻运动能力强,飞行速度快,其调控过程与代谢速度有关,需要消耗大量的能量,而能量主要产生于线粒体。线粒体一般集中在代谢活跃的区域,此外,线粒体也较为集中地分布在有较多氧化反应底物的区域(牛京京等,2011)。


图4 COG分类统计
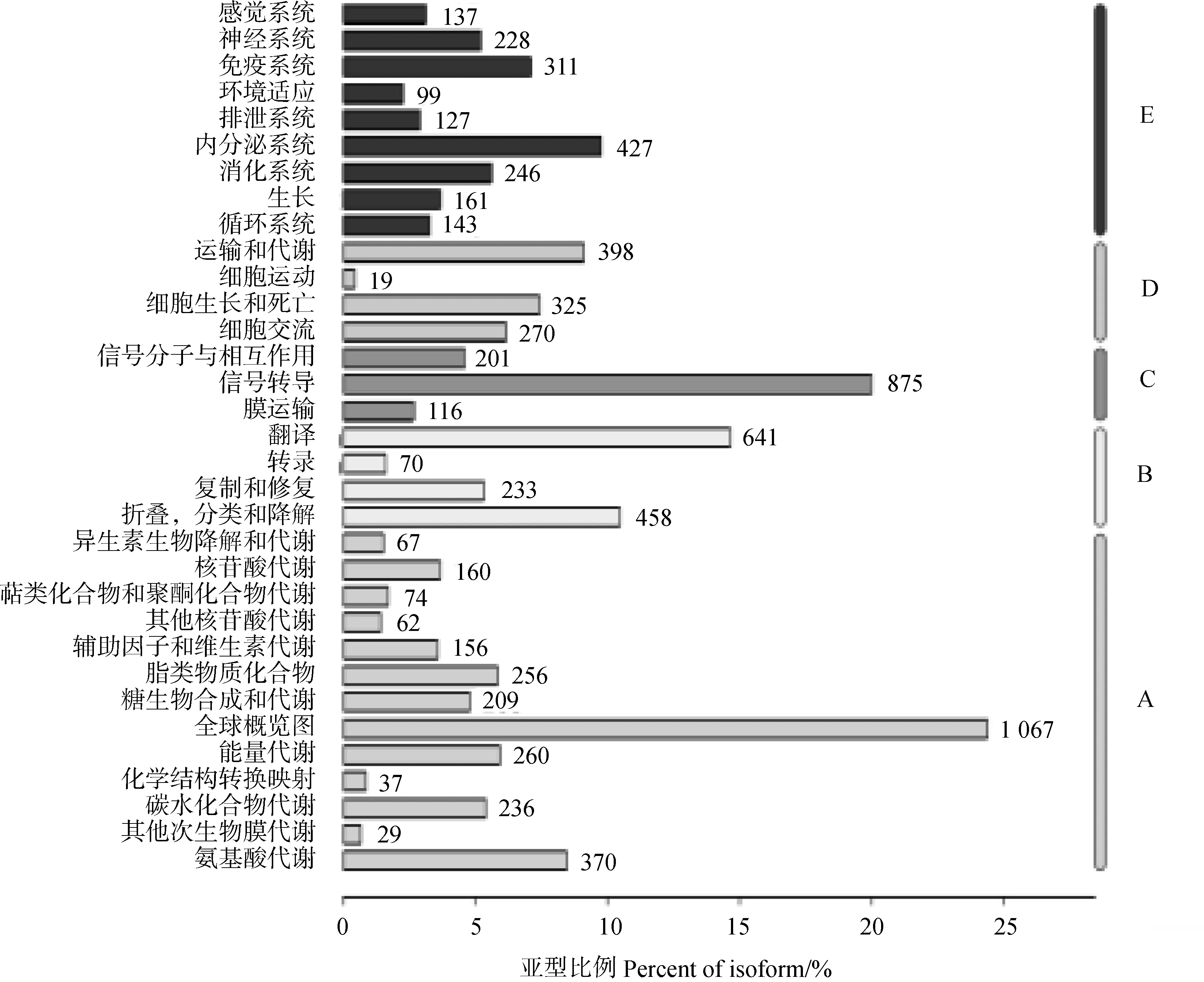
图5 KEGG 注释统计
将拼接所得的核苷酸序列用BlastX分别与NR、String、SwissProt、KEGG、Pfam数据库进行比对获得相应的注释信息,共有44 499条unigenes被注释到数据库,其中注释到Pfam数据库9 190条(20.65%),注释到KEGG数据库7 418条(16.67%),注释到String数据库5 071条(11.40%),注释到SwissProt数据库9 783条(21.98%),注释到NR数据库13 037条(29.30%)。
在我们的研究中,将核苷酸与数据库进行比对,有许多unigenes无匹配,此部分包括以下3种类型:(1)unigenes 序列片段长度过短,不能获得比对结果(张少平等,2016);(2)基因注释信息不足,近年很少有人利用转录组技术对蜻蜓进行测序分析,生物信息库还不完善,造成部分序列无法获得相应的注释结果;(3)可能有新的基因产生,在某些恶劣环境中,生物为了生存会有一些适应性的改变,相应的基因也会发生变化,在数据库中没有。
通过与各个数据库注释后获得的结果比对,找到了大量与运动有关的酶系,如微管蛋白、驱动蛋白、中间纤维、动力蛋白、肌钙蛋白、原肌球蛋白;与代谢有关的酶系,如ATP合成酶、磷酸烯醇式丙酮酸激酶、丙酮酸激酶、果糖-1,6-二磷酸激酶、己糖激酶等。但也有大量的基因没有与数据库对上,许多与运动代谢有关的酶系没有得到注释,这其间也可能有一些新出现的基因是数据库里所没有的。
动力蛋白与驱动蛋白是运动必不可少的蛋白,此次基因查询结果发现驱动蛋白(28组)、动力蛋白(27组)、原肌球蛋白(37组),有利于我们找出与运动相关基因。ATP合成酶主要参与线粒体内的氧化磷酸化过程。细胞色素c氧化亚基为生物氧化过程中的电子传递体,在酶的作用下,对组织的氧化还原有迅速的酶促作用,细胞色素c还可以在通透性增强的情况下,进入线粒体,增强细胞氧化,提高氧的利用。本研究获得19组ATP合成酶,12组细胞色素c氧化亚基和5组泛醌氧化还原酶。黄蜻的运动伴随着能量的大幅度消耗和产生,而线粒体内有关反应和酶也正是需要了解的,因此本研究结果有利于查找黄蜻所特有的与运动相关的基因,为园林害虫和农业害虫的防治提供基础。
综上所述,通过RNA-seq方法进行转录组测序所获得的与运动代谢有关的基因,为以后的相关研究提供了丰富的资源。
邓望喜. 1981. 褐飞虱及白背飞虱空中迁飞规律的研究[J]. 植物保护学报, 8(2): 74-80.
贾新平, 孙晓波, 邓衍明, 等. 2014. 鸟巢蕨转录组高通量转录组测序分析[J]. 园艺学报, 41(11): 2329-2341.
刘立春. 1994. 诱虫灯的研究进展及应用概况[J]. 华东昆虫学报, 3(1): 75-78.
牛京京, 张守纯, 金谷. 2011. 线粒体基因及其Cytb基因与昆虫分子系统学研究[J]. 生物技术通报, 4: 52-55.
吴琼, 孙超, 陈士林, 等. 2010. 转录组学在药用植物研究中的应用[J]. 世界科学技术(中医药现代化), 12(3): 457-461.
张大治, 杨贵军. 2006. 基于线粒体色素b基因序列的蜻亚科部分种类分子系统学研究(蜻蜓目:蜻科)[J]. 宁夏大学学报(自然科学版), 27(3): 255-259.
张楠, 孙桂玲, 戴均贵, 等. 2013. 银杏细胞转录组高通量测序分析[J]. 中国生物工程杂志, 33(5): 112-119.
张少平, 洪建基, 邱珊莲. 2016. 紫背天葵高通量转录组测序分析[J]. 园艺学报, 43(15): 935-946.
朱立强, 李庆花. 2016. 基于RNA-seq的杜氏盐藻全转录组测序与分析[J]. 郑州大学学报(医学版), 51(3): 289-293.
Chapman Jw, Reynolds D, Smith AD,etal. 2004. An aerial netting study of insects migrating at high altitude over England[J]. Bulletin of Entomological Research, 94(2): 123-136.
Feng HQ, Wu KM, Ni YX,etal. 2006. Nocturnal migration of dragonies over the Bohai Sea in northern China[J]. Ecological Entomology, 31: 511-520.
Freeland J, May ML, Lodge R,etal. 2003. Genetic diversity and widespread haplotypes in a migratory dragonfly, the common green darner (Anaxjunius)[J]. Ecological Entomology, 28: 413-421.
Hardy AC, Milne PS. 1938. Studies in the distribution of insects by aerial currents[J]. Journal of Animal Ecology, 7(2): 199-229.
Hobson KA, Greenberg RV, Wilgenburg SL,etal. 2010. Migratory connectivity in the rusty blackbird (Euphaguscarolinus) in North America: isotopic evidence from feathers of historical and contemporary specimens[J]. The Condor, 112: 778-788.
Hobson KA, Soto DX, Paulson DR,etal. 2012. A dragonfly (δ2H) isoscape for North America: a new tool for determining natal origins of migratory aquatic emergent insects[J]. Methods in Ecology and Evolution, 3: 766-772.
Matthews JH, Boles T, Parmesan C,etal. 2007. Isolation and characterization of nuclear microsatellite loci for the common green darner dragonflyAnaxjunius(Odonata: Aeshnidae) to constrain patterns of phenotypic and spatial diversity[J]. Molecular Ecology Notes,7:845-847.
Matthews JH. 2007. Research in motion: patterns of large-scale migration in dragonflies and birds[D]. Texas, US: The University of Texas at Austin.
1.1 一般资料 2015年—2017年应用LISS治疗复杂股骨远端骨折,男13例,女5例;年龄22~68岁,平均43岁;按AO分型:33C2型11例、33C3型7例;致伤原因:车祸伤9例,坠落伤5例,压伤4例;闭合性骨折15例,开放性骨折3例均为Gustilo Ⅱ型。手术距受伤时间5~16 d,平均7.5 d;均采用髌骨旁外侧切口,需辅助髌骨旁内侧切口3例,单纯应用LISS接骨板固定骨折14例,应用LISS接骨板结合空心螺钉或松质骨复位固定骨折4例。
Suhling F, Schenk K, Padeffke T,etal. 2004. A field study of larval develepment in a dragonfly assemblage in African desert ponds (Odonata)[J]. Hydrobiologia, 528(1): 75-85.
van Noordwijk M. 1978. A mark-recapture study of coexisting zygopteran populations[J]. Odonatologica, 7: 353-374.
Velculescu VE, Zhang L, Zhou W,etal. 1997. Characterization of the yeast transcriptome[J]. Cell, 88: 243-251.
Watanabe M, Matsuoka H, Taguchi M. 2004. Habitat selection and population parameters ofSympetruminfuscatum(Selys) during sexually mature stages in a cool temperate zone of Japan (Anisoptera: Libellulidae)[J]. Odonatologica, 33: 169-179.
Wellenreuther M, Sánchez-Guillén RA, Cordero-Rivera A,etal. 2011. Environmental and climatic determinants of molecular diversity and genetic population structure in a coenagrionid damselfly[J]. PLoS ONE, 6(6): e20440. DOI:10.1371/journal.pone.0020440.
Wikelski M, Moskowitz D, Adelman JS,etal. 2006. Simple rules guide dragonfly migration[J]. Biology Letters, 2: 325-329.
陕西省鸟类新纪录——黑翅鸢
黑翅鸢隶属于隼形目Falconiformes鹰科Accipitridae黑翅鸢属。文献曾记录中国仅有南方亚种E.c.vociferus罕见于中国南部云南、广东、广西和香港(约翰·马敬能等,2000)。但近年来该物种出现了向北扩散的趋势,分布区逐步扩大。如在福建(唐兆和等,1993;林清贤等,2004)、山东黄河三角洲(单凯等,2005)、天津(王凤琴,陈建中,2006)、北京(闻丞等,2013)、河南(卜艳珍等,2014)和四川(王疆评等,2016)等地相继作为地区新纪录发现该物种。目前已知的分布地还包括除上述地区之外的江西、江苏、上海、浙江、澳门、海南和台湾(郑光美,2011)。此次黑翅鸢在西安渭河的发现尚属首次,应为陕西省鸟类新纪录。因为个体只有1只,作者判断其为迷鸟的可能性大。该物种分布区向北甚至西北地区扩散是否与近年来的气候变化有关有待进一步研究。

图1 黑翅鸢Elanus caeruleus(王勇 摄)

图2 黑翅鸢Elanus caeruleus(廖小青 摄)
廖小凤1, 廖小青1, 于晓平2*
(1. 陕西省观鸟协会,西安710082; 2. 陕西师范大学生命科学学院,西安710119)
*通信作者, E-mail:yuxp64@163.com
Transcriptome Analysis ofPantalaflavescens(Fabricius) Based on RNA-seq
GE Yiqing, CAO Lingzhen*
(School of Life Sciences, Jiangxi Normal University, Nangchang 330000, China)
The total transcripts ofPantalaflavescens(Fabricius) were obtained by RNA-seq followed by functional analysis. The head total RNA ofP.flavescens(Fabricius) was extracted and then sequenced by Illumina platform. A total of 47 039 040 reads containing 7 102 895 040 bp were generated. There were 34 406 502 unigenes with GC content of 44.05% afterdenovoassembly. All the unigenes were annotated based on different databases, and 44 499 unigenes were annotated. In this study, all the assembled unigenes could be broadly divided into biological processes, cellular components and molecular function categories of 60 branches by gene ontology, including metabolic process, binding, catalytic activity and cellular process. Unigenes were further annotated based on COG category, which could be grouped into 26 functional categories. The results of KEGG prediction suggested that mitochondrial genes were the most abundant in the tested samples. And after KEGG ORTHOLOGY annotation, the most abundant genes were metabolic-related. The genes of mitochondria and metabolism were related with the migration mechanism ofP.flavescens(Fabricius).
Pantalaflavescens(Fabricius); transcriptome; sequencing; gene analysis; function annotation
2016-08-23 接受日期:2016-10-08
鄱阳湖湿地与流域研究教育部重点实验室(江西师范大学)开放基金项目(PK2013001); 江西省教育厅课题“基于线粒体分子标记的江西黄蜻遗传多样性研究”项目(7357)
葛怡情(1994—), 本科生, 从事动物学研究, E-mail:1757883815@qq.com
*通信作者Corresponding author, E-mail:clzclz1011@163.com
10.11984/j.issn.1000-7083.20160227
Q963
A
1000-7083(2016)06-0852-08
猜你喜欢
天天爱科学(2022年9期)2022-09-15
天天爱科学(2022年4期)2022-05-23
当代水产(2022年3期)2022-04-26
传染病信息(2021年6期)2021-02-12
航空世界(2020年10期)2020-01-19
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
财经(2017年2期)2017-03-10
财经(2016年15期)2016-06-03
财经(2016年3期)2016-03-07
