
溶剂诱导相变萃取-UPC2快速测定中成药及原料药中的香豆素
2017-01-06 04:29杜明远刘倩倩
分析测试学报 2016年12期
王 波,周 围*,冯 静,杜明远,刘倩倩
(1.甘肃出入境检验检疫局 综合技术中心,甘肃 兰州 730000;2.甘肃省检验检疫科学技术研究院,甘肃 兰州 730000)
溶剂诱导相变萃取-UPC2快速测定中成药及原料药中的香豆素
王 波1,2,周 围1,2*,冯 静1,杜明远1,刘倩倩2
(1.甘肃出入境检验检疫局 综合技术中心,甘肃 兰州 730000;2.甘肃省检验检疫科学技术研究院,甘肃 兰州 730000)
使用溶剂诱导相变萃取对中成药及原料药进行预处理,建立了中成药中香豆素的超高效合相色谱(UPC2)检测方法。样品经粉碎(60目)、乙腈-水(7∶3 ,体积比)提取,二氯甲烷诱导分相后,使用UPC2对样品中的香豆素进行检测。以3倍信噪比(S/N)确定香豆素的检出限为0.1 mg/L;线性范围为0.3~10.0 mg/L;加标回收率为89.8%~102.3%;相对标准偏差为0.31%~1.0%。同时与UPLC进行比较,结果表明该方法的选择性高,检测成本低,且分析时间仅为UPLC的1/4。该研究为高通量检测香豆素提供了一种全新的前处理及检测方法。
香豆素;溶剂诱导分相萃取;超高效合相色谱;超高效液相色谱;检测
香豆素是广泛分布于植物界中的次生代谢物质,常与生源密切的桂皮酸、黄酮类、木脂素等伴生,尤其是伞形科、芸香科、菊科、豆科、兰科和茄科等植物[1-2]。根据环上取代基及其位置的不同,常将香豆素分为简单香豆素、呋喃香豆素、吡喃香豆素和其他香豆素等。研究表明,香豆素及其衍生物除了具有抗癌、抗炎及抗凝血等生物学活性外,20世纪八九十年代,香豆素被公认为可能具有遗传毒性的致癌物质,因此欧盟规定香豆素在食品及药品中的含量不得高于2.0 mg/kg。2004年,新的研究证明,香豆素并不是遗传毒性的致癌物质,但香豆素的一小部分成分具有肝毒性,在此基础上,欧盟食品安全管理局公布了香豆素的日服用限量为0~0.1 mg/kg(按体重计算)[3-6]。为确保以肉桂及桂枝为中药原料制成的中成药顺利出口及合理制定每日的口服剂量,对香豆素的检测显得尤为重要。
香豆素的测定方法一般有液相色谱法[1,7-10]、液相色谱-串联质谱法[11-12]、气相色谱-串联质谱法[13-14]、紫外分光光度法[15]等,但紫外分光光度法测定香豆素的方法选择性较差,易受其它成分的干扰,所需试剂较多、操作繁琐。而对于液相色谱及串联质谱,除了实验成本较高外,有机溶剂的使用还会对环境造成污染。样品预处理是中成药及其原料等复杂基质中分离检测目标物的重要环节。溶剂诱导相变萃取法已成功用于血浆样品中痕量药物的测定[16]。该方法与传统液-液萃取法相比,具有操作简单快速等优点,且新形成的有机相与常用的反相色谱流动相兼容,可直接进样分析,无需在进样前进行溶剂转化。通过文献查阅,该样品处理方法尚未见用于中成药中香豆素测定的报道。
超临界流体(Supercritical fluid,SCF)是指物质高于其临界温度和临界压力时的一种物态,它既不是气体,也不是液体,但兼有气体的低粘度,液体的高密度,以及介于气液体之间的扩散系数等特征。超高效合相色谱(Ultraperformance convergence chromatography,UPC2)是以超临界流体为流动相,依靠流动相的溶剂化能力进行分离分析的一种分析技术;其在传统超临界流体色谱(Supercritical fluid chromatography,SFC)的原理上,应用超高效液相色谱(UPLC)的硬件技术,并使用更小的色谱柱填料(≤ 2 μm)达到快速、高效的分离[17-19]。SFC和UPLC的结合,克服了传统SFC在实验过程中精密度差、重现性低的缺点,在整个操作过程中有机溶剂使用量少,是一种绿色的分离技术。目前采用UPC2对香豆素进行快速检测的方法尚未见报道。
本实验通过建立一种新的中成药前处理方法,即溶剂诱导相变萃取,利用分相后的高度选择性对复杂基质中的香豆素进行检测;文中还系统考察了UPC2对香豆素分离的影响因素,并与UPLC进行比较,相关研究为中成药及其原料药中香豆素的提取、分离及检测提供新的思路。
1 实验部分
1.1 仪器与试剂
超高效合相色谱仪(美国Waters公司)配二极管阵列检测器,Waters Empower 3数据处理系统;移液枪(10~100 μL,100~1 000 μL,美国Thermo Electron公司);分析天平;50 mL聚乙烯管。
香豆素(Dr.Ehrenstorfer公司,纯度99%);CO2(中国汇能公司,纯度99.997%);乙腈、甲醇(德国Merck公司);蒸馏水(屈臣氏);其余试剂均为分析纯。
1.2 标准溶液配制
标准贮备液:准确称取10.00 mg香豆素,用乙腈-水(7∶3)定容至100 mL容量瓶中,配制成100 mg/L的香豆素标准溶液。4 ℃下冷藏待用。
标准工作液:准确移取5.0,2.5,625,313,156,52 μL标准贮备液于50 mL容量瓶中,乙腈-水(体积比7∶3)准确定容,分别配制成10.00,5.00,1.25,0.62,0.31,0.10 mg/L的标准工作液。4 ℃下冷藏待用。
1.3 色谱条件
色谱柱:UPC2CSH Fluoro-Phenyl(2.1 mm×150 mm,1.7 μm);流动相A为CO2;B为含0.2%甲酸的甲醇溶液;流速0.5 mL/min,进样体积1.0 μL,柱温50 ℃,检测波长278 nm,动态背压(ABPR)为1 900 psi。梯度洗脱:0~3.0 min,98%~90% A;3.0~4.5 min,90%~80% A;4.5~5.5 min,80%~70% A;5.5~6.0 min,70%~98% A;6.0~8.0 min,98%A平衡2 min。
1.4 样品来源
不同种类的中成药(T1~T5)及原料药均由某制药厂提供,所有样品粉碎后待用。
1.5 样品预处理
称取0.50 g(精确至0.01 g)粉碎后的样品于50 mL聚乙烯管中,准确添加35 mL乙腈-水(7∶3)提取液,涡旋2.0 min,超声提取15 min后准确加入5.0 mL二氯甲烷,涡旋2.0 min,4 ℃条件下13 000 r/min冷冻离心10 min。离心完毕后,溶液分层,上层为乙腈层,下层为水层,取乙腈层溶液1.5 mL,过0.22 μm有机相膜,待测定。
2 结果与讨论
2.1 实验条件的优化
2.1.1 提取溶剂及提取条件的选择 为了完全提取中成药及原料药中的香豆素,本实验称取同一中成药样品适量,料液比为1∶50(g/mL),分别加入50%,60%,70%,80%及90%的乙腈水溶液进行超声提取。结果表明,当使用70%乙腈-水溶液对样品进行超声提取时,待测样品中香豆素的浓度最高,为376.17 mg/L。固定提取溶剂不变,考察了超声提取时间(5,15,25,35 min)和提取次数(1,2,3,4次)以及料液比(1∶30,1∶50,1∶70,1∶90)对香豆素提取率的影响。结果表明,在最佳提取条件下(即提取溶剂为70%乙腈水溶液,料液比为1∶70,超声15 min提取1次),样品中香豆素的提取效率最高,提取浓度达到414.85 mg/L。
2.1.2 诱导分相剂的选择 有文献报道,几乎所有与乙腈互溶的疏水性溶剂都能作为诱导分相剂诱导乙腈-水体系分层[16],因此,本实验选择具有代表性的不含氧诱导剂如二氯甲烷、无机盐如氯化钠及含氧诱导剂如正辛醇对香豆素标准溶液(1.25 mg/L)进行诱导分相萃取,比较了水层和乙腈层中香豆素的含量,并通过计算回收率确定香豆素的转移率。结果显示,采用正辛醇作为诱导分相剂进行提取时,目标物的回收率较低,仅为74.2%,使用二氯甲烷和氯化钠进行诱导分相,香豆素的回收率均在95%以上,表明香豆素已高选择性地转移至乙腈层中。此外,使用氯化钠作为诱导分相剂时,水层高浓度的盐不利于UPC2对水层化合物的分析。故实验选择二氯甲烷作为诱导分相剂。
2.1.3 色谱柱的选择 由于中成药及原料药中的成分极其复杂,色谱柱对目标物的保留、峰形及选择性起着至关重要的作用。样品按“1.5”处理后,使用3种不同的色谱柱BEH(3.0 mm×100 mm,1.7 μm),BEH 2-EP(2.1 mm×150 mm,1.7 μm)及CSH Fluoro-Phenyl(2.1 mm×150 mm,1.7 μm)对样品乙腈层中的香豆素进行分离检测。结果表明,当使用BEH及BEH 2-EP色谱柱时,受杂质的干扰,严重影响香豆素定量的准确性,采用CSH Fluoro-Phenyl色谱柱时,乙腈层中的香豆素无任何杂质的干扰。故本实验选择的色谱柱为CSH Fluoro-Phenyl柱。
2.1.4 流速的选择 使用超临界CO2作为流动相时,相比传统的正相及反相色谱的流动相,它具有低粘度和高扩散系数,以及较小的粘度,从而减小了分离过程中的阻力。在相同的色谱柱条件下,可使用较高的流速。考虑到流速主要对分析时间和系统压力产生影响,本实验在0.3,0.5,0.8,1.0 mL/min范围内进行考察,在保证理想的分析时间和系统压力的前提下,最终选择0.5 mL/min为最佳流速。
2.1.5 助溶剂的选择 以CO2超临界流体为流动相时,通常对极性化合物的洗脱能力较弱,可通过加入甲醇、乙醇、乙腈、异丙醇及挥发性酸或盐作为助溶剂来增加对极性化合物的洗脱能力,以适应对不同极性目标化合物的分离,并得到较好的峰形及理想的保留时间。本实验考察了甲醇、乙腈两种最为常见的助溶剂对香豆素的分离效果。结果显示,使用乙腈作为助溶剂时,峰形拖尾严重,且保留时间达10 min ,使用甲醇作为助溶剂时,香豆素的保留时间为2.69 min,但香豆素仍有拖尾,而通过在甲醇加入0.2%甲酸,可得到较好的峰形,保留时间稍有提前(2.47 min),故选择含0.2%甲酸的甲醇溶液作为助溶剂。
2.1.6 动态背压(ABPR)的选择 超临界流体在不同压力下有不同的溶解能力,其溶解能力随压力的增加而增加,故压力是影响分离过程的重要因素之一。动态背压(ABPR)可在整个运行过程中维持CO2的超临界流体状态,实验考察了ABPR在1 600,1 700,1 900,2 000 psi条件下对样品分离的影响。结果表明:随着背压升高,系统压力随之升高,系统超临界流体溶解度的增加使得香豆素的保留时间减少。当背压为1 900 psi时,香豆素与杂质得到很好分离,且保留时间短、色谱峰形对称,故选择背压为1 900 psi。
2.1.7 色谱柱温度的选择 在超临界流体状态下,SCF的溶解能力随温度的增加而降低。实验考察了30,40,50,60 ℃的条件下对目标物分离的影响。结果表明,随着温度的升高,香豆素的保留时间逐渐增加,在此前提下,综合考虑样品中杂质与香豆素的分离程度以及每个样品的分析时间,本实验选择50 ℃为最佳分离温度。
2.2 UPC2的方法学考察
2.2.1 稳定性实验 准确称取编号为T4的中成药样品0.50 g,按“1.5”方法处理样品,在“1.3”色谱条件下,分别在0,4,12,24,48 h进样6次,T4样品中香豆素的峰面积相对标准偏差(RSD)分别为0%,0.38%,0.44%,0.61%,0.69%和0.66%。表明样品溶液在48 h内稳定。
2.2.2 专属性实验 取标准溶液、阴性供试品溶液以及中成药样品的乙腈层溶液,按“1.3”色谱条件进样分析并比较,结果显示,香豆素的出峰位置无杂质干扰,表明该方法的专属性较好。
2.2.3 线性范围及定量下限 按“1.2”分别配制成10.00,5.00,1.25,0.62,0.30,0.10 mg/L的标准工作液。按照“1.3”色谱条件,每个浓度进样1.0 μL,以浓度(x,mg/L)为横坐标,峰面积(y)为纵坐标,进行线性回归。结果表明,香豆素在0.3~10.0 mg/L范围内线性关系良好,线性方程为y=153.8x+8.74;根据信噪比(S/N)为3和10确定香豆素的检出限和定量下限分别为0.1 mg/L和0.3 mg/L。
2.2.4 精密度实验 按照“1.3”所述色谱条件,对添加高、中、低3个不同浓度(10.00,1.25,0.30 mg/L)香豆素标准溶液的中成药样品重复进样6次,测得日内精密度的RSD为0.31%~0.63%;日间精密度的RSD为0.49%~0.92%,可以看出该方法对检测中成药及原料药中的香豆素具有较好的日间和日内精密度,能满足对香豆素的检测要求。
2.2.5 重复性实验 准确称取同一中成药样品,按“1.5”方法进行样品处理,“1.3”色谱条件进样分析,计算香豆素的含量及RSD。6份同一中成药样品中香豆素含量的RSD为1.0%,表明方法的重复性较好。
2.2.6 加标回收率 准确称取同一中成药样品,分别加入高、中、低3个不同浓度(10.00,1.25,0.30 mg/L)的香豆素标准溶液,在优化条件下进样分析。结果显示,3个不同浓度的香豆素加标回收率为89.8%~102.3%,表明本方法准确可靠。
2.3 与UPLC方法的比较
按照“1.5”对样品处理后,采用相同色谱柱CSH Fluoro-Phenyl(2.1 mm×150 mm,1.7 μm),使用UPLC仪进样2.0 μL进行分析,流动相为0.2%甲酸甲醇和水溶液,梯度洗脱,278 nm下检测。另取相同的供试品溶液按“1.3”色谱条件分析,分别得到香豆素标准溶液的UPC2和UPLC色谱图(见图1)。由图可以看出,UPC2与UPLC相比,因为流动相性质及保留机理的不同,香豆素标准溶液在UPC2的出峰时间(2.15 min)较UPLC的出峰时间(8.52 min)缩短了近4倍。由此可见,本实验建立的UPC2快速检测中成药中香豆素的方法与传统液相色谱方法相比,既节省溶剂,降低检测成本,又能进行快速分离,实现高通量检测。

2.4 实际样品的测定
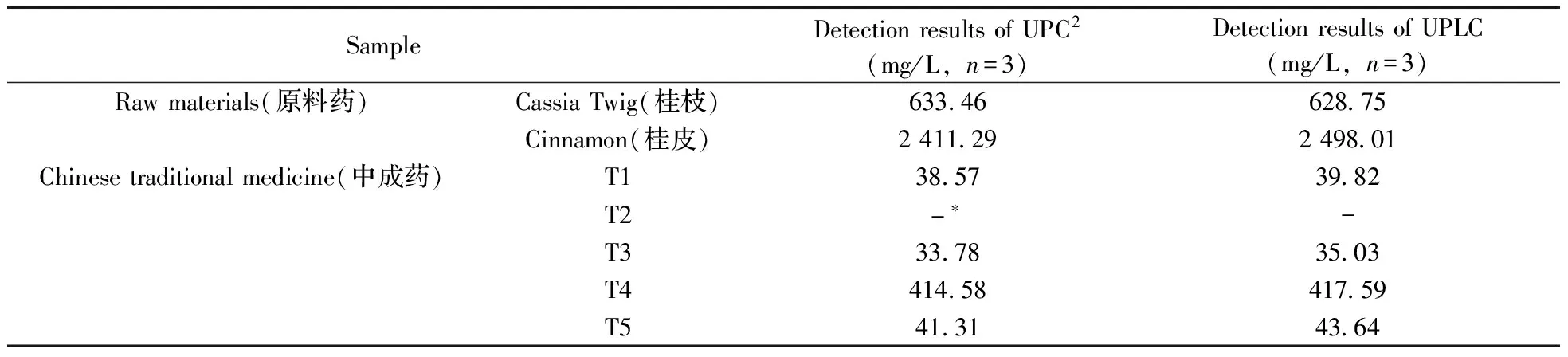
取5批中成药及2批原料药样品经粉碎后(60目),准确称取0.50 g,按“1.5”进行样品处理,在“1.3”色谱条件下进样分析,图2为样品T4总提液、乙腈层以及水层的色谱图;每个样品重复3次,通过光谱图确定峰纯度,外标法计算香豆素的含量,样品中香豆素的含量结果见表1。由表1可知,除样品T2未检出香豆素外,其余中成药均检出香豆素,且T4样品的浓度最高,达到414.58 mg/L;对于原料药的分析结果显示,肉桂中香豆素的浓度为2 144.29 mg/L,约为桂枝的4倍。此外,通过UPC2和UPLC对样品检测结果的比较,表明UPC2的检测结果除了与UPLC具有相当的准确度外,检测时间仅为UPLC的1/4,故使用UPC2可对香豆素进行快速准确、高通量的检测。

SampleDetectionresultsofUPC2(mg/L,n=3)DetectionresultsofUPLC(mg/L,n=3)Rawmaterials(原料药)CassiaTwig(桂枝)6334662875Cinnamon(桂皮)241129249801Chinesetraditionalmedicine(中成药)T138573982T2-∗-T333783503T44145841759T541314364
*no detected
3 结 论
本文采用二氯甲烷作为诱导剂对样品中的香豆素进行萃取,香豆素可高选择性地存在于乙腈层中,并且在萃取过程中无需特殊装置,操作简便,是一种稳定可靠的分析方法。同时,首次采用UPC2建立了检测中成药及原料药中香豆素的方法,方法学考察结果显示,该方法的灵敏度高,重现性、稳定性良好,检测时间短,为中药复杂体系的色谱分析提供了新的方向。通过与UPLC比较,分析时间仅为UPLC的1/4,可节约大量的分析时间,提高检测效率。采用所建立的最佳色谱方法对5批中成药及2批原料药材进行了测定,实验结果显示肉桂和桂枝中香豆素的含量较高,为确保以肉桂及桂枝为中药原料制成的中成药顺利出口及合理制定每日口服剂量提供了数据支持。
[1] Li J,Li N,Wu T,Wang C.LifeSci.Instrum.(李洁,李楠,武婷,王超.生命科学仪器),2005,3(5):34-37.[2] Zheng L,Zhao T,Sun L X.LISHIZHENMed.Mater.Med.Res.(郑玲,赵挺,孙立新.时珍国医国药),2013,24(3):714-716.
[3] Li J Z,Huang C S,Liu H X,Chen X H.J.GuangxiTeachersEduc.Univ.:Nat.Sci.Ed.(李锦周,黄初升,刘红星,陈希慧.广西师范学院学报:自然科学版),2007,24(1):94-96.
[4] Zhang S Y,Meng L,Gao W Y.Chin.J.Chin.Mater.Med.(张韶瑜,孟林,高文远.中国中药杂志),2005,30(6):410.
[5] Kong L L,Hu J F,Chen N H.Chin.Pharmacol.Bull.(孔令雷,胡金凤,陈乃宏.中国药理学通报),2012,28(2):165-169.
[6] Ballin N Z,Sørensen A T.FoodControl,2014,38(4):198-203.
[7] Xi H W,Ma Q,Wang C,Bai H,Liu X,Wang Y.J.Instrum.Anal.(席海为,马强,王超,白桦,刘茜,王烨.分析测试学报),2010,29(12):1168-1172.
[8] Ma H J,Ma Q,Li W T,Meng X S,Li J R,Bai H,Jiao Y,Zhang X L.J.Chin.Chromatogr.(马会娟,马强,李文涛,孟宪双,李晶瑞,白桦,焦阳,张晓丽.色谱),2013,31(5):416-422.
[9] Zhao X Y,Fu X F,Wang P,Li J,Hu X Z.J.Anal.Sci.(赵晓亚,付晓芳,王鹏,李晶,胡小钟.分析科学学报),2011,27(1):49-52.
[10] Jin M C,Chen X H,Li X P.Chin.J.Chromatogr.(金米聪,陈晓红,李小平.色谱),2007,25(2):214-216.
[11] Sua J,Zhang C,Zhang W,Shen Y H,Li H L,Liu R H,Zhang X,Hu X J,Zhang W D.J.Chromatogr.A,2009,1216(11):2111-2117.
[12] Yang R J,Wei B W,Gao H,Yu W J.Chin.J.Chromatogr.(杨荣静,卫碧文,高欢,于文佳.色谱),2012,30(2):160-164.
[13] Song G S,Xu Y M,Zhou L,Zhang L T.ActaPharm.Sin.(宋更申,徐艳梅,周丽,张兰桐.药学学报),2016,51(4):626-630.
[14] Zhao X Y,Lin Y F,Hu X Z,Wang P,Fu X F,Li J.Chin.J.Anal.Lab.(赵晓亚,林雁飞,胡小钟,王鹏,付晓芳,李晶.分析试验室),2010,29(3):76-79.
[15] Luo X R,Long Q D,Yang J,Tian Y F.GuangdongAgric.Sci.(罗喜荣,龙庆德,杨军,田弋夫.广东农业科学),2012,39(6):98-99.
[16] Zhang M S,Liu G Z,Li S J,Chen B,Yao S Z.Chem.J.Chin.Univ.(张鸣珊,刘国柱,李圣军,陈波,姚守拙.高等学校化学学报),2010,31(8):1517-1521.
[17] Liu Q Q,Zhou W,Wang B,Wang L T,Yang S X.FoodSci.(刘倩倩,周围,王波,王丽婷,杨盛鑫.食品科学),2014,35(16):208-211.
[18] Zhou W,Wang B,Liu Q Q,Yang S X,Wang L T.Chin.J.Anal.Chem.(周围,王波,刘倩倩,杨盛鑫,王丽婷.分析化学),2015,(1):115-120.
[19] Samokhin A S,Revelsky I A,Chepelyansky D A,Parenago O O,Pokrovsky O I,Lepeshkin F D,Ustinovich K B,Revelsky A I.Russ.J.Phys.Chem.B,2013,6(7):769-785.
Rapid Determination of Coumarin in Chinese Traditional Patent Medicine and Raw Materials by UPC2and Solvent Induced Extraction
WANG Bo1,2,ZHOU Wei1,2*,FENG Jing1,DU Ming-yuan1,LIU Qian-qian2
(1.Central Laboratory of Technical Center of Gansu Entry-Exit Inspection and Quarantine Bureau,Lanzhou 730000,China;2.Gansu Province Inspection and Quarantine Science and Technology Research Institute,Lanzhou 730000,China)
A method of solvent induced phase extraction and ultra performance convergence chromatography(UPC2) was established for the determination of coumarin in Chinese traditional patent medicine and raw materials.Samples were crushed(60 mesh),and extracted with acetonitrile-water(7∶3,by volume) using dichloromethane as induced separation solvent.The targeted compound was detected by UPC2method.The limit of detection(LOD,S/N=3) for coumarin was 0.1 mg/L.The linear range of coumarin was in the range of 0.3-10.0 mg/L.The spiked recoveries ranged from 89.8% to 102.3% and the relative standard deviations were between 0.31% and 1.0%.Compared with UPLC method,the method showed the advantages of high selectivity and low detection cost,and its analysis time is only 1/4 of the UPLC.This study provided a new pretreatment and detection method for the high throughput detection of the coumarin.
coumarin;solvent induced extraction;ultra performance convergence chromatography(UPC2);ultra performance liquid chromatography(UPLC);detection
2016-05-18;
2016-06-29
10.3969/j.issn.1004-4957.2016.12.019
O657.72;TQ460.72
A
1004-4957(2016)12-1622-06
*通讯作者:周 围,博士,研究员,研究方向:色谱分析、食品药品安全及检测,Tel:0931-8513950,E-mail:zhouwei845@163.com
猜你喜欢
中老年保健(2022年3期)2022-08-24
云南化工(2021年7期)2021-12-21
中国生殖健康(2020年5期)2021-01-18
农药科学与管理(2019年8期)2019-11-23
中国盐业(2018年20期)2019-01-14
天然产物研究与开发(2018年10期)2018-11-06
中国生殖健康(2018年5期)2018-11-06
天然产物研究与开发(2018年6期)2018-07-09
天然产物研究与开发(2018年2期)2018-04-04
中华皮肤科杂志(2014年4期)2014-12-19
