
解读WHO(2017)头颈部肿瘤分类
——口咽肿瘤
2017-03-20 03:48方三高魏建国周晓军
临床与实验病理学杂志 2017年12期
方三高,魏建国,周晓军
WHO(2017)头颈部肿瘤分类[1](简称新版)与WHO(2005)头颈部肿瘤病理学和遗传学分类[2](简称旧版)相比,将口咽从传统口腔中独立出来,集中描述,体现了12年来该领域研究的最新进展[3-5],强调了FISH和(或)PCR技术直接检测HPV的重要性,但允许使用免疫组化标记p16,作为间接测试HPV状态可靠的替代品,使分子分型变得简单易行。基于HPV测试状态,新版将口咽鳞状细胞癌(oropharyngeal squamous cell carcinomas, OPSCC)分为HPV阳性及HPV阴性,而非传统分类的角化型与非角化型。如同子宫颈癌,已经明确HPV阳性的OPSCC同样由高危型HPV感染引起,但实践中不再推荐对其进行组织学分级。精准分类有助于肿瘤界定、解释流行病学趋势及预测临床预后,使诊断的可重复性与临床一致性更强。本文将重点介绍新增病种及其相关背景的知识。
1 WHO头颈部肿瘤分类表和TNM分期
分类表详见WHO(2017)头颈部肿瘤分类[6]。新版所附为第7版,目前AJCC/UICC已推出TNM分期第8版[7-8]。
2 口咽癌解剖学基础、流行病学、病原学及病因学
2.1口咽解剖学基础新版口咽部分包括舌根、扁桃体及腺样体,继“口腔与可移动舌部肿瘤”之后,作为第5章,成为并列关系;而旧版口咽被置于“口腔”项下,为包含属性。口腔本身由唇、牙龈、磨牙后三角、硬腭、颊黏膜、舌体和口底构成;而口咽由腭扁桃体、软腭、舌根、后轮廓乳头和咽后壁组成。尽管口腔和口咽均被覆复层鳞状上皮,形成一个连续的腔室或穹窿,但口咽存在大量的扁桃体组织,高度特化的淋巴上皮(即网状上皮)衬覆于扁桃体隐窝,为HPV感染及其驱动肿瘤的发生提供了更加宽松的环境[3]。解剖上,口咽位于口腔后方,上临软腭,下以会厌顶部的假想水平线为界,前靠咽峡,后抵舌后1/3,侧缘由咽腭弓和腭扁桃体构成,后壁含腺样体或咽扁桃体,而一般临床上所谓的扁桃体指的是腭扁桃体,左右各一,大部分由排成小结节或滤泡的淋巴样组织构成,位于前后咽柱之间的扁桃体沟(隐窝三角)内,从软腭一直延续到舌背部,表面呈脑回状,有许多隐窝或裂隙几乎延伸到腺体全层,无输入淋巴管和包膜下窦,此处的鳞状细胞癌向下浸润可达深部组织,如舌根、咽侧壁或喉咽,向上可累及腭与鼻咽部。舌的咽部不可动,简称舌根,其下有淋巴样组织形成的舌扁桃体,表面呈圆凸形,亦可见小涎腺组织。而咽鼓管扁桃体为咽鼓管咽口附近黏膜内的淋巴组织,经耳咽管开口进入鼻咽后方,这些较大的淋巴组织团块呈环状排列形成咽淋巴环,淋巴瘤好发,而恶性上皮性肿瘤也并非罕见。
2.2流行病学与口腔癌一样,口咽部癌同样以鳞状细胞癌最常见,既往认为与长期吸烟、酗酒、嚼烟、咀嚼槟榔等密切相关,近30年流行病学研究发现,尽管男性吸烟量明显降低,但OPSCC发病率不减反增,原因在于无症状性高危型HPV隐性感染成为主要传染源,可经口交、舔阴、舔肛、接吻等亲密行为接触性传播[9]。作为一种常见的性传播疾病(sexually transmitted disease, STD),从传染病流行的基本环节分析,高危型HPV完整病毒颗粒为传染源,经皮肤黏膜密切接触传染为传播途径,40~59岁的白人男性为易感人群,他们往往不吸烟,但具有较高的社会经济地位及较多的性伴侣。
2.3病原学超过90%的OPSCC病例由HPV16感染引起,HPV16基因结构详见图1[10]。HPV属于小分子DNA病毒,无包膜,其裸露的核壳体直径约55 nm,衣壳有72个颗粒,构成20面对称球体。HPV由核酸和衣壳蛋白组成双链环状DNA,以共价闭合的超螺旋结构、开放的环状结构及线性分子3种形式存在,约7 900个碱基对,基因组编码9个开放读码框架(open reading frame, ORF),分为3个编码区:早期区(early region, ER)、晚期区(later region, LR)和长控区(long control region, LCR)。LCR也叫上游调节区(upstream regulatory region, URR),属于非编码区(non-coding region, NCR),含有多个结合位点,通过与转录因子的相互作用,调节ER基因的转录。HPV16具有2个启动子元件:早期启动子(early promoter, PE, 也称为p97)和晚期启动子(late promoter, PL,也称为p670),分别表示病毒转录物5′端位点与RNA起始位点,上皮分化期间调控mRNA的剪接与表达。早期多聚腺苷酸化和晚期多聚腺苷酸化[10],分别位于nt4215和nt7321,表示基因组内早期和晚期多聚腺苷酸化位点的位置。ER包括E1~E8区,其中E6、E7为两个主要致癌基因,其作用及功能状态变化详见图2。
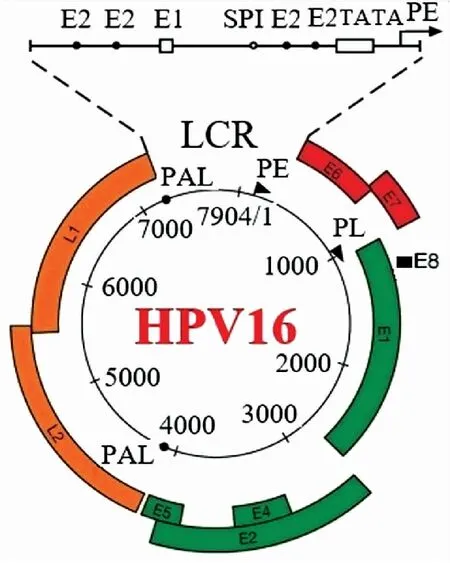
图1 HPV16基因结构示意图[10]
2.3.1HPV16基因主要成分的作用 ER区中E6通过抑制p53而阻断凋亡,引起细胞无限增生并向恶性转化。E7通过抑制pRB而使细胞周期失控而发生永生化。E1涉及病毒DNA复制,在病毒起始复制中起关键作用。E8^E2由小的E8部分和E2C结构域组成,由较不保守的“铰链区”高危型和高度保守的DNA结合结构域(DNA binding domain, DBD)组成。E8^E2蛋白以不同于其他早期病毒基因的方式调控病毒的复制与转录[11-12]。E2负性调节E6和E7,维持凋亡和细胞周期的调控。整合时,病毒破坏E2的结构,使之失活,直接导致E6和E7过度表达。E4与胞质蛋白成熟有关,结合并破坏宿主细胞骨架,促进病毒颗粒释放,形成挖空细胞外观。E5位于E2下游,通过细胞生长因子受体激活上调控。晚期转录区组成病毒的衣壳,且与病毒的增殖有关,包括L1:主要衣壳蛋白,是疫苗的主要成分,L2:次要衣壳蛋白,是市售抗体的主要识别位点。长控区:URR或LCR,调节基因转录。
2.3.2E6、E7的致癌作用 如图2[13]所示,细胞内E6蛋白与泛素连接酶E6相关蛋白(E6-AP)形成复合物,特异性地结合p53,通过抑制p53而阻断细胞凋亡。E7蛋白是HPV的主要转化蛋白,与控制细胞周期有关的肿瘤抑制蛋白视网膜母细胞瘤蛋白(RB)亲和力极高,E7与RB结合,打断了RB与E2F1的结合,使E2F1游离出来,从而发挥其转录因子的作用,使细胞周期由G1期向S期转化,导致细胞周期失控而发生永生化。E6、E7过度表达,使p53对细胞生长负调节功能丧失,从而失去对细胞周期的正常调控,同时抑制免疫系统的应答,引起免疫逃逸,造成细胞无限增生并导致人类染色体端粒体酶逆转录酶基因(hTERC)扩增,维持端粒长度,细胞永生,免于凋亡而向恶性转化。
根据E6、E7和L1基因序列可对HPV进行分型,序列与已知HPV基因型同源性超过90%的属于同一个基因型,基因型可以进一步分为亚型,如果与原型有超过98%的同源性,认为是同一亚型;如果与原型相似性在90%~98%之间,认为是一种新的变异型。迄今为止,已经描述了85种HPV基因型和120多个分离株(可能为新的基因型)。感染人类的HPV共40种,其中13~15种属于高危型,如HPV16型及18型,4~6种为低危型,如HPV6型及11型,一般仅检测高危型,市售HPV E6, E7 mRNA检测试剂盒,阳性结果提示HPV病毒曾经感染宿主,临床上可以利用不依赖于病理材料的液体活检,经血清学检测并预测预后[14],一般认为阳性者预后较好。HPV仅在人类上皮细胞中才能增殖,在具有一定分化程度的角质形成细胞内复制,主要在终末分化的上层细胞中进行病毒基因组扩增及病毒颗粒成熟与组装,无法体外培养,病毒不进入血液,不产生病毒血症。80%口咽部HPV为“一过性”感染,大多数在1年内被清除;20%属于持续性感染。存在两种形式,一种是病毒DNA借助于HPV受体α6β4整合素整合到宿主细胞基因组中[15-16];另一种是病毒DNA游离于其外。

图2 E6和E7在HPV相关性肿瘤细胞周期途径中的作用及基因功能状态变化示意图[13]
2.4病因学自1983年[17]首次提出并于1985年发现[18]以来,现已明确高危型HPV是HPV阳性的OPSCC的病因和生物学致癌物。病毒DNA整合到宿主染色体中,被认为是致癌作用的重要驱动因素[15-16]。相关肿瘤进展的关键事件包括持续感染、基底上皮细胞中病毒早期基因的失调表达、局部免疫抑制和染色体改变的积累[19-20]。皮肤及黏膜中朗格汉斯细胞(Langerhans, LC)起源于树突状细胞(dendritic cell, DC),为主要的抗原递呈细胞(antigen presenting cell, APC),正常情况下与其他细胞协同作用,为皮肤及黏膜提供免疫监视,但HPV感染可导致皮肤及黏膜中LC明显减少,造成局部免疫抑制,引起宿主对病毒耐受,在其他致癌因素协同作用下导致鳞状细胞癌的发生。
2.5病理学扁桃体具有特殊的非角化上皮形态,其隐窝上皮富于网状结构,HPV可自上而下,然后自下而上形成病毒循环[21]:首先病毒颗粒通过微小破损经胞吞作用感染上皮基底层细胞,其表面的受体与病毒结合,使病毒黏附于细胞表面,进入胞质后通过不明机制被传送到核,随着基底层干细胞样细胞的分裂以及纵向逐步分化成熟,病毒复制并组装,成熟的病毒在表面细胞分离时释放到环境中。在此循环过程中E4具有协同作用,其高表达使胞质破坏、凹陷,形态上表现为挖空细胞,促进病毒释放而易于播散。在基底层细胞中,病毒的复制处于非产生病毒颗粒阶段,以低拷贝附加体形式维持游离低拷贝量的DNA,而在分化的基底层上层细胞中,病毒转换了复制模式,可扩增至数千份,合成衣壳蛋白,组装游离释放病毒颗粒,产生新的病毒体。
参与细胞周期进程的细胞蛋白表达由p16INK4a调节,通过抑制Cyclin D/CDK的活性,参与负反馈环路。在未感染上皮中,E2F1的释放依赖外部生长因子刺激Cyclin D/CDK的活性,以允许pRb磷酸化及E2F1释放;而在高危型HPV感染通路中,E7蛋白可靶向降解pRb,使转录因子家族成员E2F1释放,结果促使基底细胞和副基底细胞进入S期。与此同时,高危型E6蛋白通过蛋白酶体途径介导的泛素化和降解,直接调节并抑制细胞中p53的水平。在未感染的细胞中p53能够维持在低水平,部分是由于MDM2的正常活性所致,MDM2即鼠双微体2(murine double minute 2),也叫E3泛素-蛋白连接酶,MDM2癌蛋白泛素化并拮抗p53,是p53肿瘤抑制因子的重要负性调控因子(图3)。

图3 高危型HPV感染干扰调节上皮分化和细胞增殖的分子通路示意图[21]
黄色显示S期进展所必需的上调基因,包括p16与Ki-67等细胞周期G1、S、G2、M时相中存在多个调节转换的关键节点,其中以G1→S期最为重要,细胞一旦从G1期进入S期,就不再依赖外界信息刺激,而很快自动完成分裂过程。G1节点是分水岭,将决定细胞是继续增殖,还是进入G0期或离开细胞周期进入终末分化或死亡。细胞周期节点的转换受控于“细胞周期蛋白-细胞周期依赖性激酶-细胞周期依赖性激酶抑制因子”这些分子网络,其中研究最为广泛的一组是:Cyclin D1/CDK4/p16[22]。如图3示,p16在细胞周期G1节点中与Cyclin D1竞争结合CDK4并特异性抑制其活性,组织细胞尤其是带有损伤DNA的细胞进入S期,从而控制细胞分裂[22]。正常的p53蛋白具有促进细胞凋亡及DNA修复功能,使细胞静息于G1期,而在DNA损伤或缺氧时活化,使依赖p53的周期素依赖激酶抑制者p21和DNA修复基因上调性转录,细胞在G1期出现生长停滞,进行DNA修复,如修复成功,细胞进入S期;如修复失败,则通过活化bax基因使细胞进入凋亡,以保证基因组的遗传稳定。故正常的p53蛋白又被称为“分子警察”。而低危型HPV的E6蛋白不能与p53结合,不影响p53的稳定,其E7基因产物与pRB的亲和力低,致癌性极低。
p16INK4a抗体简写为p16,简单而便宜,临床上用于间接检测HPV。在高级别鳞状上皮内病变中p16的表达模式为大块阳性,即核与胞质弥漫性连续着色;而在低级别鳞状上皮内病变中完全不表达或仅局灶阳性。p16与p53、Cyclin D1及Ki-67等一起标记并相互补充,增加可信度,同时能与鳞状上皮化生鉴别。p16INK4a的过度表达是由于Rb的丧失而发生的,可以被用作HPV阳性OPSCC的诊断替代标志物。图4显示了p16过表达的分子机制。正常细胞周期受周期素Cyclin D1和CDK的正性调节,在G1期,Cyclin D1和CDK4或CDK6结合后,磷酸化灭活pRb,释放转录因子ELF1,促进细胞由G1期进入S期,促进细胞增殖。同时p16等几个CDK抑制基因对细胞周期进行负调节,其产物p16蛋白可与Cyclin D1竞争性结合CDK4或CDK6,并特异性抑制CDK4活性,使之不能磷酸化pRb,阻止细胞进入S期,从而形成了p16INK4a-Cyclin D-CDK4-pRb精细调节级联,在此级联中,pRb-ELF1复合物也抑制基因p16本身的转录。首先,E6蛋白与宿主细胞的p53结合,E6AP引起细胞p53蛋白降解,损害细胞凋亡,使细胞永生化。其次,E7蛋白结合并灭活Rb蛋白,Rb调节E2F1的活性,通过钝化Rb,增加E2F1的水平,促进细胞周期进程。CDKN2A编码可变剪接的p14ARF和p16INK4a基因。p14蛋白抑制MDM2泛素化p53。p21诱导的p21和p16均通过抑制Rb磷酸化,促进细胞周期进程中的细胞周期蛋白形成,而Rb反馈抑制p16产生。
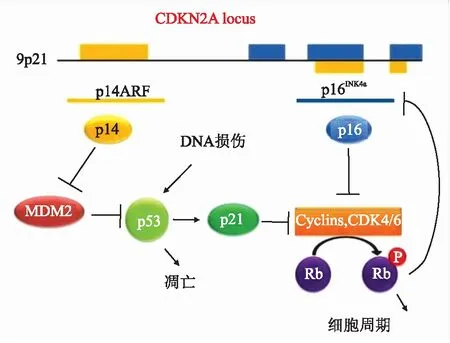
图4 CDKN2A基因产物和p53调控[22]
表观遗传学被定义为不改变DNA序列、研究基因表达及遗传变化的领域,包括DNA甲基化、组蛋白翻译后修饰、小RNA及非编码RNA(small RNA and non-coding RNA, sRNA/ncRNA)等。研究显示,头颈部鳞状细胞癌基因突变包括CCND1扩增、CDKN2A(p16)突变、缺失及高甲基化、RB1突变和缺失、E2F1扩增、MYC扩增和TP53扩增。其中基因CCND1变化最频繁,几乎每一个HPV阴性肿瘤均有CDKN2A的失活,大多数均失活了TP53。图5为OPSCC表观遗传途径汇总[23],显示了导致与致癌作用相关途径的基因表达失调情况,其中凋亡途径受p53等固有信号及细胞表面受体外在信号双重调节。DNA损伤、紫外线辐射或钙离子内流等多重信号可引起p53反应而触发凋亡信号通路,导致细胞色素C从线粒体释放出来,引起酶联反应,启动程序性细胞死亡程序。通过细胞表面死亡受体Fas等,激活胞质中类似含半胱氨酸的天冬氨酸蛋白水解酶(Caspase)的一组蛋白酶,活化下游相应Caspase结构,触发细胞凋亡。高危型HPV融合到人类基因组中,导致E6和E7高表达,引起OPSCC中p53、Rb和Polycomb抑制复合物(包括EZH2、SUZ12、EED和HOXD)相关途径与下游表观遗传学调控变化。FOXM1和HOTAIR被推定为基于口腔鳞状细胞癌(oral cavity squamous cdl carcinoma, OCSCC)研究而在OPSCC中发挥作用。除了DNA甲基化调控外,HPV基因组的转录尚需后期REA剪切和多聚腺苷酸酸化作用才能成熟。
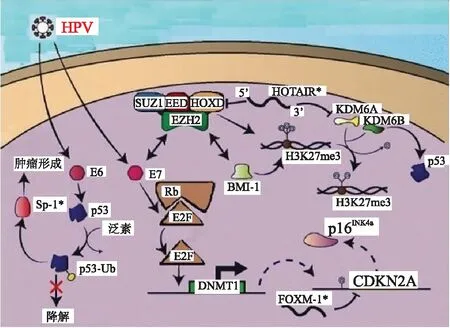
图5 口咽部鳞状细胞癌表观遗传学汇总图[23]
3 口咽癌诊断、治疗及预防
WHO分类如同总纲,纲举则目张。作为金标准,形态学分类、分型及分级有助于精准诊断或个体化处理,可借助Ki-67、p16、p53及E6、E7等[24]检测增加准确性,为此获得详细而完整的资料显得非常重要。临床应进行彻底的口腔内外检查,识别可疑病变,及时进行穿刺切取或切除活检,以明确诊断,特别是对初始提示性症状如持续性喉咙疼痛、吞咽困难、声音嘶哑、耳痛、淋巴结肿大或体重减轻应引起临床高度重视,进一步进行医学评估和随访。尽管新技术层出不穷[25],但检查与治疗原则应遵循美国国立综合癌症网络(National Comprehensive Cancer Network, NCCN)指南及最新指南。为了早期预防,有人建议使用针对HPV16和HPV18感染的预防性HPV疫苗,可能会减少OCSCC及OPSCC的发病率[26]。
4 新增病种
4.1鳞状细胞癌,HPV阳性HPV阳性的OPSCC具有独特的统计学、遗传学、临床病理及预后特征[27]。一般经历高危型HPV持续感染-免疫逃逸-病毒整合-病毒癌基因表达-E6/E7癌基因介导细胞转化-肿瘤抑制因子p53和pRB失活-侵袭性癌基因转变-恶性肿瘤形成,这样一个连续而漫长过程[26]。通常认为HPV阳性的鳞状细胞癌起源于扁桃体隐窝,这种特殊上皮始终保持不成熟、非角化和基底样外观,与表面上皮的异型增生无关,几乎不存在原位癌[28],肿瘤常形成较大的实性巢团或呈小叶状生长,具有推进式光滑边界,很少或无间质反应,但常伴淋巴细胞浸润。癌细胞呈长梭形,角化通常不存在或程度有限,形态也不如角化型那样肥胖,胞质相对稀少,导致核质比高,圆形或椭圆形的核深染或嗜双色,核仁不明显,核分裂及凋亡/坏死常见。若弥漫性表达p16,WHO建议使用简洁、直观而清晰的命名“鳞状细胞癌,HPV阳性”;对未进行HPV检测直接或p16标记,而仅有典型HPV感染相关形态,新版推荐使用“鳞状细胞癌,未检测HPV,形态高度提示HPV关联”这样的描述性术语。新版将非角化型鳞状细胞癌定义为具有成熟的鳞状分化,比例小于整个肿瘤表面积的10%。50%~55%的OPSCC和约70%的HPV阳性OPSCC属于非角化型,此外,HPV阳性OPSCC的其他形态还包括乳头状鳞状细胞癌、腺鳞癌[包括最近描述的“纤毛状腺鳞癌(ciliated adenosquamous carcinoma, CASC)”],淋巴上皮样癌(未分化癌)及肉瘤样(梭形细胞)癌,其共同特征是p16弥漫或大块阳性,Ki-67高表达,增殖指数通常>70%。临床上肿瘤多为Ⅲ或Ⅳ期,尽管形态学为低分化或未分化,但约80%对治疗反应良好,生存率高于HPV阴性组。
4.2鳞状细胞癌,HPV阴性经检测,未发现HPV感染或不表达p16的OPSCC,相应地被命名为“鳞状细胞癌,HPV阴性”[1]。肿瘤往往表达p53,常伴角化,与HPV阳性的鳞状细胞癌相比,患者平均发病年龄大6岁,更具遗传多样性,预后较差[1,5,27]。50%~63%的头颈部鳞状细胞癌发生p53错义突变,导致稳定蛋白质的关键结合功能丧失,甚至以显著负性调节方式使残余野生型p53失活。HPV阳性的鳞状细胞癌与吸烟及TP53突变有关,但HPV感染和p53基因突变可能是OPSCC发生过程中的共存分子事件。
4.3小细胞癌与鼻腔鼻窦命名为“神经内分泌癌”不同,口咽部直接命名为小细胞癌。尽管文献报道例数不多[29],但发病率持续上升,有证据表明HPV阳性的OPSCC可以经历小细胞癌转化,呈高核质比及核铸型。如同肺及其他部位的同名肿瘤一样,口咽部小细胞癌与吸烟、高级别细胞特征、神经内分泌标志物表达、临床侵袭性如广泛扩散和极差的生存率密切相关,与非小细胞癌形成鲜明对比,故新版将其分开作为一个独特病种,而不只是HPV阳性口咽癌的一种变异形式。镜下细胞呈实性片层状密集排列,胞质稀少,圆形或短梭形裸核,染色质细腻,核仁不明显。常见血管浸润及坏死。可出现腺样或鳞状细胞分化,但不到肿瘤体积的10%。注意活检时当旁边的血管等未被压扁清晰显示而肿瘤细胞呈弥漫蓝染一片(蓝湖),呈人工挤压或形成“掐丝样结构”(烂糊),恰好就是小细胞癌的特点,而非贸然建议临床重取活检,但需借助于免疫组化与嗅神经母细胞瘤、淋巴瘤、Ewing肉瘤/PENT、胚胎性横纹肌肉瘤等蓝色小圆细胞肿瘤鉴别[1,29]。
4.4多形性腺癌(polymorphousadenocarcinoma,PMA) 由旧版“多形性低级别腺癌(polymorphous low grade adenocarcinoma, PLGA)”取消其限定词“低级别”并与“小涎腺筛状癌(cribriform adenocarcinoma of minor salivary glands, CAMSG)”合并而来。更名后新版归于一组异质性肿瘤,通常属于低级别,极少数可发生高级别转化,临床呈侵袭性。PLGA约60%发生于腭部,临床上形成无痛性肿块,偶见出血、毛细血管扩张或黏膜溃疡形成。曾定义为“以细胞学一致性、形态学多样性、浸润性生长、低转移潜能为特征的涎腺恶性上皮性肿瘤”。大部分呈结节性生长,但缺乏包膜,肿瘤组织形成条索、腺管或筛状巢团,有时具有束状和靶状特征,伴蓝灰色间质,核卵圆形,淡染,形态温和。10%~33%的患者可局部复发,多达9%~15%出现淋巴结转移,但无远处转移并导致患者死亡的报道[30]。而CAMSG为单形性肿瘤,具有更广泛的筛状结构及巢周间隙,呈“肾小球样”外观,单个肿瘤细胞圆形或椭圆形,轮廓不规则,染色质细腻,貌似乳头状甲状腺癌,但不表达TG与TTF-1。舌筛状腺癌(cribriform adenocarcinoma of tonge, CACT)可能为PLGA的一个亚型,绝大多数发生于舌,通常在舌根部形成肿块,可转移至颈部淋巴结,但无远处转移[31]。CACT发病年龄25~70岁(平均50.4岁),无性别差异。肿瘤沿黏膜下生长,被纤维结缔组织分隔成小叶状,呈实性或筛状排列,一些实性岛中同样可见特征性的肾小球样结构,中央为宽的微滤泡性乳头,借较窄的缝隙与周围一层柱状细胞分开。偶有肿瘤细胞呈梭形并可见少许导管。细胞核大小一致,染色浅,常有重叠,这种透明状细胞核,易与甲状腺乳头状癌混淆。核分裂少见,无明显的出血和坏死。间质透明样变,偶见砂砾体,可见浸润性边缘,累及周围软组织。鉴别诊断:多形性腺瘤(pleomorphicadenoma, PA)由增生的上皮和肌上皮及软骨样基质构成,尽管可呈多结节状生长,但界限清楚,包膜完整,缺乏浸润,而PLGA缺乏包膜呈浸润性生长,没有PA良性浆细胞样肌上皮细胞,黏液软骨样区域不明显,可出现筛状或肾小球样结构。与腺样囊性癌(adenoid cystic carcinoma, AdCC)的鉴别主要根据细胞形态,PMA的细胞为立方或柱状,核泡状,胞质常嗜酸,缺乏筛状假囊性腔隙及AdCC中基底样细胞特征,AdCC中几乎不出现乳头和肾小球样外观,缺乏细胞多形性和高核分裂活性。
4.5CASC口咽部腺鳞癌被认为是鳞状细胞癌的一种罕见变异型,更具临床侵袭性[32]。早在2004年,Alos等[33]概述了AsqCA诊断标准,即表面鳞状上皮异型增生/原位癌,浸润性生长模式,明显角化,离散腺癌灶(通常在肿瘤的深部),缺乏中间型细胞和明显的核非典型性。镜下鳞状细胞癌当中可见形态良好的癌性腺体,形成圆润、“冲压”状腔隙,周围可见局灶性嗜碱性小球和嗜酸性黏蛋白。腺鳞两种成分细胞均可表达p16,属于HPV相关性肿瘤。其他部位的腺鳞癌不管以何种成分为主,鳞状细胞癌和腺癌需各占10%以上。新版未对CASC的腺体比例设定最低要求,认为腺癌的数量与疾病复发或生存无关。口咽部腺鳞癌大部分为非角化型鳞状细胞癌,但有的呈囊性变、腺体形成及黏蛋白产生,部分肿瘤细胞顶端呈条索状,形成特征性的纤毛,故名。
[1] El-Naggar A K, Chan J K C, Grandis J R,etal. WHO classification of tumors of head and neck tumours[M]. 4th ed. Lyon: IARC Press, 2017:1-347.
[2] Barnes L, Eveson J W, Reichart P,etal. WHO pathology and genetics classification of tumors of head and neck tumours[M]. 3rd ed. Lyon: IARC Press, 2005:1-430.
[3] Westra W H, Lewis J S Jr. Update from the 4th edition of the World Health Organization classification of head and neck tumours: oropharynx[J]. Head Neck Pathol, 2017,11(1):41-47.
[4] Gondim D D, Haynes W, Wang X,etal. Histologic typing in oropharyngeal squamous cell carcinoma: a 4-year prospective practice study with p16 and high-risk HPV mRNA testing correlation[J]. Am J Surg Pathol, 2016,40(8):1117-1124.
[5] Fakhry C, Westra W H, Wang S J,etal. The prognostic role of sex, race, and human papillomavirus in oropharyngeal and nonoropharyngeal head and neck squamous cell cancer[J]. Cancer, 2017,123(9):1566-1575.
[6] 方三高. WHO(2017)头颈部肿瘤分类[J]. 诊断病理学杂志, 2017,26(9):638-641.
[7] Brierley J D, Gospodarowicz M K, Wittekind C. TNM classification of malignant tumours[M]. 8th ed. Toronto: Wiley Blackwell, 2016:1-272.
[8] Mizumachi T, Homma A, Sakashita T,etal. Confirmation of the eighth edition of the AJCC/UICC TNM staging system for HPV-mediated oropharyngeal cancer in Japan[J]. Int J Clin Oncol, 2017,22(4):682-689.
[9] Taberna M, Inglehart R C, Pickard R K,etal. Significant changes in sexual behavior after a diagnosis of human papillomavirus-positive and human papillomavirus-negative oral cancer[J]. Cancer, 2017,123(7):1156-1165.
[10] Doorbar J, Quint W, Banks L,etal. The biology and life-cycle of human papillomaviruses[J]. Vaccine, 2012,30(Suppl 5):F55-F70.
[11] Straub E, Dreer M, Fertey J,etal. The viral E8^E2C repressor limits productive replication of human papillomavirus 16[J]. J Virol, 2014,88(2):937-947.
[12] Lace M J, Anson J R, Thomas G S,etal. The E8--E2 gene product of human papillomavirus type 16 represses early transcription andreplication but is dispensable for viral plasmid persistence in keratinocytes[J]. J Virol, 2008,82(21):10841-10853.
[13] Hayes D N, Van Waes C, Seiwert T Y. Genetic landscape of human papillomavirus-associated head and neck cancer and comparison to tobacco-related tumors[J]. J Clin Oncol, 2015,33(29):3227-3234.
[14] Nelson H H, Pawlita M, Michaud D S,etal. Immune response to HPV16 E6 and E7 proteins and patient outcomes in head and neck cancer[J]. JAMA Oncol, 2017,3(2):178-185.
[15] Walline H M, Komarck C M, McHugh J B,etal. Genomic integration of high-risk HPV alters gene expression in oropharyngeal squamous cell carcinoma[J]. Mol Cancer Res, 2016,14(10):941-952.
[16] Aksoy P, Abban C Y, Kiyashka E,etal. HPV16 infection of HaCaTs is dependent on β4 integrin, and α6 integrin processing[J]. Virology, 2014,449(1):45-52.
[17] Syrjänen K, Syrjänen S, Lamberg M,etal. Morphological and immunohistochemical evidence suggesting human papillomavirus (HPV) involvement in oral squamous cell carcinogenesis[J]. Int J Oral Surg, 1983,12(6):418-424.
[18] Löning T, Ikenberg H, Becker J,etal. Analysis of oral papillomas, leukoplakias, and invasive carcinomas for human papillomavirus type related DNA[J]. J Invest Dermatol, 1985,84(5):417-420.
[19] Groves I J, Coleman N. Pathogenesis of human papillomavirus-associated mucosal disease[J]. J Pathol, 2015,235(4):527-538.
[20] Doorbar J. Model systems of human papillomavirus-associated disease[J]. J Pathol, 2016,238(2):166-179.
[21] Doorbar J, Egawa N, Griffin H,etal. Human papillomavirus molecular biology and disease association[J]. Rev Med Virol, 2015,25 (Suppl 1):2-23.
[22] Guo T, Califano J A. Molecular biology and immunology of head and neck cancer[J]. Surg Oncol Clin N Am, 2015,24(3):397-407.
[23] Lindsay C, Seikaly H, Biron V L. Epigenetics of oropharyngeal squamous cell carcinoma: opportunities for novel chemotherapeutic targets[J]. J Otolaryngol Head Neck Surg, 2017,46(1):9-10.
[24] Holzinger D, Wichmann G, Baboci L,etal. Sensitivity and specificity of antibodies against HPV16 E6 and other early proteins for the detection of HPV16-driven oropharyngeal squamous cell carcinoma[J]. Int J Cancer, 2017,140(12):2748-2757.
[25] Dias F L, Walder F, Leonhardt F D. The role of transoral robotic surgery in the management of oropharyngeal cancer[J]. Curr Opin Oncol, 2017,29(3):266-271.
[26] Yang A, Farmer E, Wu T C,etal. Perspectives for therapeutic HPV vaccine development[J]. J Biomed Sci, 2016,23(1):75-76.
[27] D'Souza G, Westra W H, Wang S J,etal. Differences in the prevalence of human papillomavirus (HPV) in head and neck squamous cell cancers by sex, race, anatomic tumor site, and HPV detection method[J]. JAMA Oncol, 2017,3(2):169-177.
[28] Black C C, Ogomo C. Does pTis exist in HPV-driven tonsillar carcinomas? An ultrastructural review and examination of two cases[J]. Ultrastruct Pathol, 2017,41(1):55-61.
[29] Misawa K, Kawasaki H, Matsuo R,etal. Human papillomavirus-associated small cell carcinoma/neuroendocrine carcinoma of the oropharynx: a report of two cases[J]. Springerplus, 2016,5(1):1847-1848.
[30] Xu B, Aneja A, Ghossein R,etal. Predictors of outcome in the phenotypic spectrum of polymorphous low-grade adenocarcinoma (PLGA) and cribriform adenocarcinoma of salivary gland (CASG): a retrospective study of 69 patients[J]. Am J Surg Pathol, 2016,40(11):1526-1537.
[31] Michal M, Denisa K, Dmitry V,etal. Cribriform adenocarcinoma of the tongue and minor salivary glands: a review[J]. Head Neck Pathol, 2013,7(Suppl 1):3-11.
[32] Radkay-Gonzalez L, Faquin W, McHugh J B,etal. Ciliated adenosquamous carcinoma: expanding the phenotypic diversity of human papillomavirus-associated tumors[J]. Head Neck Pathol, 2016,10(2):167-175.
[33] Alos L, Castillo M, Nadal A,etal. Adenosquamous carcinoma of the head and neck: criteria for diagnosis in a study of 12 cases[J]. Histopathology, 2004,44(6):570-579.
猜你喜欢
中华养生保健(2020年7期)2020-11-16
健康必读·下旬刊(2018年4期)2018-06-04
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中华老年多器官疾病杂志(2016年7期)2016-04-28
中国卫生标准管理(2015年25期)2016-01-14
听力学及言语疾病杂志(2015年5期)2015-12-24
医学研究杂志(2015年5期)2015-06-10
医学研究杂志(2015年5期)2015-06-10
中国医药科学(2015年15期)2015-02-27
现代检验医学杂志(2015年5期)2015-02-06
