
不同茂金属化合物结构优化分析*
2017-11-03 02:33,,
合成材料老化与应用 2017年5期
, ,
(中国石油兰州化工研究中心,甘肃兰州 730060)
不同茂金属化合物结构优化分析*
王海,张鹏,李忠
(中国石油兰州化工研究中心,甘肃兰州 730060)
以Cp2ZrCl2(二氯二茂锆)为基准茂金属化合物,采用DFT(密度反函数理论)对不同烷基引入环戊二烯基时形成的配体结构进行优化,并对优化过程中产生的差异进行分析。实验结果表明:相对于Cp2ZrCl2的能量变化,正丁基引入环戊二烯基时能量变化降低,其他烷基引入时能量变化升高;乙基引入时产生的位移及梯度最小,而正丁基最大;正丁基引入环戊二烯基时结构优化速度快于乙基的引入,结构收敛性好。
茂金属化合物,密度反函数理论,烷基
茂金属是至少含有一个环戊二烯基或取代的环戊二烯基配体的一大类金属有机配合物。1951年,Miller[1]和Pauson[2]首次合成了二茂铁化合物。1952年,Wilkinson[3]和Fisher[4]揭示了二茂铁的夹心状结构,这一发现有力地推动了金属有机化学的发展。根据茂金属的数量和结构特征,一般将用作催化剂的茂金属配合物分为无桥二茂金属、桥联二茂金属、单茂金属以及具有刚性结构的单茂基金属化合物。至今研究得最多的用于烯烃聚合的茂金属催化剂是ⅣB族过渡金属的茂金属,其中锆的二茂基配合物和钛的单茂基配合物活性很高,已在聚烯烃工业化生产中得到实际应用。
已有多个研究组对茂金属的活性中心的形成及催化剂机理做过系统的研究。I.Tritto等[5]采用1H NMR和13C NMR波普直接跟踪Cp2ZrCl2(二氯二茂锆)与MAO(甲基铝氧烷)的反应过程及其产物结构,发现MAO有很强的烷基化作用,Cp2ZrCl2与MAO反应首先生成烷基化的茂金属,然后生成阳离子活性中心。T.Ziegler等人用基于密度泛函理论(DFT)的分子模拟方法对Cp2ZrCl2/MAO催化乙烯聚合的链增长过程进行了理论计算和分析,得出的结论与α-agostic效应的链增长模型一致[6]。
本文采用DFT对不同茂金属化合物进行模拟计算,分析不同茂金属化合物成键步骤存在的差异,特别是优化步骤时产生的能量变化、最大梯度、最大位移量。
1 实验
1.1 原材料
所选的茂金属化合物,Alfa Aesar,a Johnson Matthey Company生产,纯度在97%。采用的DFT软件为Material Studio 5.5。
1.2 模拟细节
采用DFT软件中Dmol3模块对茂金属化合物结构进行模拟,任务栏选择:能量;功能参数选择GGA的PBE;性能选择:电荷分布。
2 结果与讨论
2.1不同茂金属化合物成键优化时能量及能量变化
以Cp2ZrCl2为基准化合物,采用DFT模拟不同茂金属化合物成键步骤,产生的总能量及能量的变化如表1所示。
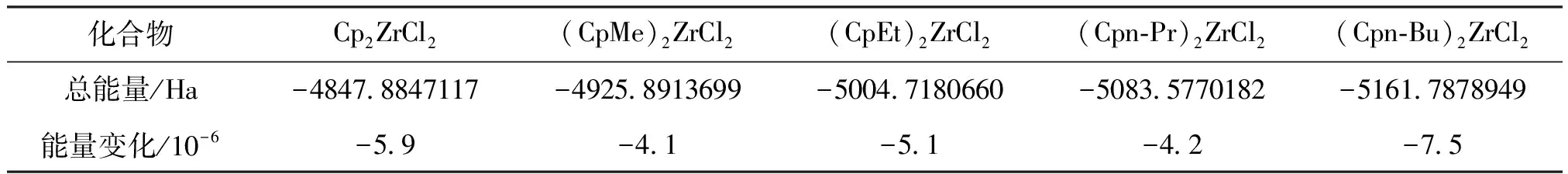
表1 不同茂金属化合物成键总能量及能量变化Table 1 The total bond energy and energy change of different metallocene compounds
从表1中可见,总能量随着取代基的增大而减小,说明随着环戊二烯基上引入烷基,化合物成键更加容易;能量变化值随着取代基的增大而增大,直至正丁基取代基的引入促使能量变化值减小,说明正丁基的引入很大程度上改变了配体的电荷密度,并且与其他三种取代基表现出不同的趋势效应。
2.2 不同茂金属化合物成键梯度及位移量变化
以Cp2ZrCl2为基准化合物,采用DFT对不同茂金属化合物成键时的梯度值及产生的最大位移进行模拟分析,结果如表2所示。
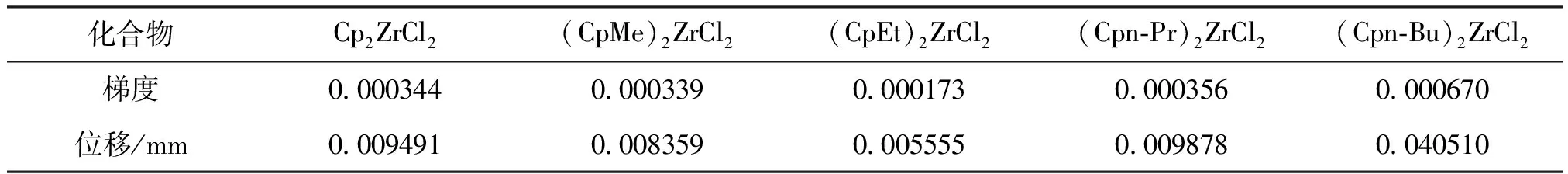
表2 不同茂金属化合物成键梯度及位移Table 2 Bond gradient and displacement of different metallocene compounds
从表2中可见,乙基引入环戊二烯基时产生的梯度值最小,产生的位移最小,说明采用DFT对该化合物进行成键优化时结构收敛性差,优化路径较难;正丁基引入环戊二烯基时产生的梯度值最大,产生的位移最大,说明采用DFT对化合物该化合物进行成键优化时结构收敛性好,优化路径容易。
2.3 不同茂金属化合物收敛性分析
为了进一步分析乙基与正丁基引入环戊二烯基时产生的巨大差异,采用DFT对两种茂金属化合物成键时的收敛性进行对比分析,结果如图1、图2所示。
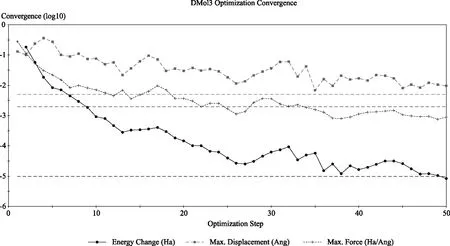
图1 (CpEt)2ZrCl2 结构收敛模拟图Fig.1 Structural convergence simulation of (CpEt)2ZrCl2 metallocene compound
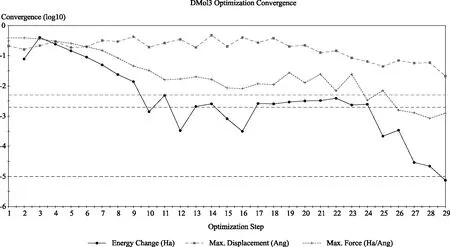
图2 (Cpn-Bu)2ZrCl2 结构收敛模拟图Fig.2 Structural convergence simulation of (Cpn-Bu)2ZrCl2 metallocene compound
从图1和图2可见,正丁基取代环戊二烯基成键时几何优化在29步内完成,乙基取代环戊二烯基成键时几何优化在43步内完成,正丁基明显比乙基收敛速度快。
3 结论
(1)以Cp2ZrCl2为基准化合物,正丁基引入环戊二烯基成键时能量变化降低,其他烷基引入环戊二烯基成键时能量变化增大。
(2)乙基引入环戊二烯基时梯度及位移值最小,正丁基引入时梯度及位移值最大。
(3)正丁基相比于乙基引入环戊二烯基时几何优化步骤快,结构收敛性好。
[1] Miller S A,Tebboth J A,Tremaine J F. J. Chem. Soc.,1952:632.
[2] Kealy T J,Pauson P J. Nature,1951,168:1039.
[3] Wilkinson G,Rosenblum M,Whiting M C,et al. J. Am. Chem. Soc.,1952,74:2125.
[4] Fisher E O. Angew. Chem.,1952,22:620.
[5] Tritto I,sacchi M C,Li S. Macromol. Chem.,Rapid Commum.,1994,15:217.
[6] Lohrenz J C W,Woo T K,Fan L Y,et al. J. Organomet. Chem.,1995,497:91.
OptimizingAnalysisoftheStructuresofDifferentMetalloceneCompounds
WANG Hai,ZHANG Peng,LI Zhong
(Lanzhou Petrochemical Research Center,Lanzhou 730060,Gansu,China)
The metallocene compounds which based on Cp2ZrCl2(dichloro zirconocene),could optimizing the structures the ligands,which is formed by introduction of cyclopentadienyl groups to the different alkyl groups by DFT (density inverse function theory),and analysing the differences in the optimization process. The experimental results showed that when n-butyl was introduced into cyclopentadienyl groups,the energy change was lower than that of Cp2ZrCl2,and the energy change was increased when the other alkyl groups were introduced. Furthermore,the displacement and gradient were the least by introducing the ethyl groups,while the situation of n-butyl groups were opposite. When the n-butyl groups were introduced into cyclopentadienyl,the structural optimization speed was faster than that of ethyl groups,and the structural convergence was better.
metallocene compounds,DFT,alkyl
TQ 325.1+4
中石油科技管理部项目(2011A-2104)
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
太原理工大学学报(2022年2期)2022-03-21
化工环保(2021年3期)2021-06-17
原子与分子物理学报(2020年5期)2020-03-17
天然产物研究与开发(2018年8期)2018-09-10
考试周刊(2018年39期)2018-04-19
中国资源综合利用(2016年7期)2016-02-03
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
