
发作性共济失调一例及文献复习
2018-04-26 03:32郭荣静常婷刘煜林宏
中国神经免疫学和神经病学杂志 2018年2期
郭荣静 常婷 刘煜 林宏
发作性共济失调(episodic ataxia,EA)是一种常染色体显性遗传、离子通道突变所致的神经肌肉疾病,临床上主要表现为头晕、恶心、呕吐,以及发作性小脑性共济失调的表现等。1946年Parker首次报道了11例发作性共济失调患者,依据基因定位和临床特点分为2类:(1)发作性共济失调Ⅰ型(episodic ataxiatype 1,EA1);(2)发作性共济失调Ⅱ型(episodic ataxiatype 2,EA2)。随着对疾病的认识以及家族基因连锁分析,2007年Jen等[1]将其分为以下几类:即EA1-EA6以及其他,临床上最常见的仍为EA1和EA2,其在多个种族以及家族均有报道。同年Kevin等[2]报道了定位于染色体19q13的发作性共济失调家系,考虑为EA7;2014年Conroy等[3]报道了EA8。由于EA发病率低,临床特征不典型,极易被误诊。本文报道1例临床考虑为EA的患者,并复习相关文献,对EA进行了系统的总结分析,期望对临床诊疗该病有所帮助。
1病例报告患者男性,38岁,主因“发作性头晕2个月”入唐都医院神经内科。患者入院2个月前主要表现为头晕以及步态不稳,如喝醉酒样,严重时有恶心以及欲跌倒感、步态不稳、视物模糊、心慌,偶有言语含糊不清,无复视、耳鸣听力下降,无吞咽困难、饮水呛咳,无肌肉跳动、头痛、肢体麻木无力,发作与头位以及体位改变无关,间隔约1~2周发作1次,每次持续3~4 h后症状完全缓解,发作间期完全正常,于外院(具体时间不详)查头颅MRI未见异常,在外院以及作者医院心内科按照“眩晕综合征”治疗后症状未见改善,后自行就诊于作者医院门诊收入作者科室诊治。
患有癫痫失神发作17年,平时服用“卡马西平 0.1 g 3次/d、丙戊酸钠 0.2 g 3次/d以及苯妥英钠 0.1 g 3次/d”,癫痫控制不佳,仍有间断2个月发作1次。
入院后查体未见明显异常;完善头颅磁共振血管成像(MRA)、24小时动态脑电图、心电图、心脏彩超、眼震电图未见异常,血常规、空腹血糖、肝肾功能及电解质等均在正常参考值范围,苯妥英钠、卡马西平以及丙戊酸钠血药浓度均在正常参考值范围内。住院期间患者无明确诱因发作,自诉头晕以及行走不能。发作当时查体:言语含糊不清,双眼向注视侧可见粗大水平眼震,四肢肌力Ⅴ级,深浅感觉均正常,双侧指指试验、跟膝胫试验欠稳准,如醉酒样不能行走;症状约持续0.5 h后缓解,3 h时完全好转。住院期间无特殊治疗,患者病情平稳。出院后患者规律服用乙酰唑胺 25 mg 2次/日,后多次电话访视1年,上述症状未再发作。
2讨论各型EA临床特点见表1。
2.1EA1
2.1.1发病机制:KCNA1是目前惟一被公认与EA1有关的基因[4],位于染色体12p13,在高度保守区,只有一个外显子,编码电压依赖性钾离子通道Kv1.1亚单位。Kv1.1可表达在中枢神经系统(小脑、海马及皮层)和周围神经,维持神经元动作兴奋性,动作电位的产生、传导等。该基因突变大部分表现为EA1,发病率为1∶500 000,目前发现有30多个突变位点,多为错义点突变,最近发现其可影响RNA编辑[5]。但在双胞胎中发现,即使同一位点突变,临床表现和疾病发生概率也可不同[6],这意味着该病可能存在不全外显率。
2.1.2临床表现: EA1发作期主要表现为四肢及躯干共济失调,同时可有言语含糊和持续性的肌纤维颤搐、神经性肌强直等表现。肌纤维颤搐多发生在眼周及手指。EA1患者也可以出现无力、眩晕、视物模糊,少数患者可伴有癫痫,发作类型为强直阵挛发作、部分性发作或部分性发作继发全身发作,发作时患者因感到严重失去平衡而想坐下或躺下来。一些患者合并有平衡和运动协调障碍、发育迟缓、舞蹈手足徐动症、腕痉挛、认知和言语功能障碍、肌张力增高、小腿肥大、呼吸暂停、口唇发紫、呼吸困难、骨骼肌畸形等。
上述发作症状多在儿童或青少年时期出现,诱因包括物理或情感创伤,以及某些刺激如发烧、惊吓、急剧的运动、前庭热刺激、焦虑、重复的屈膝、摄入咖啡因、旋转木马、高温[7]等。症状多持续数秒钟到数分钟不等,但在少数情况下可持续数小时;发病频率不定,可每日发作15次左右,有些患者发生频率更低,可1个月1次。发作可能随着年龄增长而消失,患者常被误诊为癫痫或心因性疾病。近年有回顾性研究发现EA1患者中至少有1/5小脑症状及体征持续存在[8]。
2.1.3检查以及治疗: EA1患者在发病期肌电图可见肌纤维颤搐,发作间期基本正常,诱发小腿缺血以及过度换气时肌电图可出现异常。发病期和间期影像学检查均正常[9]。
乙酰唑胺可以减少EA1发生的频率和减轻症状,其具体机制尚不明确,目前多考虑为该药可以调节细胞内pH值和HCO3-离子梯度,同时还可以影响脑脊液pH值。因此,对乙酰唑胺敏感的患者有可能出现碱中毒。其他不良反应有多汗、肾结石、感觉异常、皮疹、无力以及胃肠不适,禁用于肝、肾以及肾上腺功能不全的患者[10]。硫噻嗪、卡马西平[11]同样可以改善EA1症状,感觉异常及间断的腕痉挛是硫噻嗪的不良反应。苯妥英钠可以减少共济失调和肌纤维抽搐的发生,同时改善肌强直和运功功能,因为其可导致永久性小脑功能障碍和肌肉萎缩,故年轻患者应慎用。
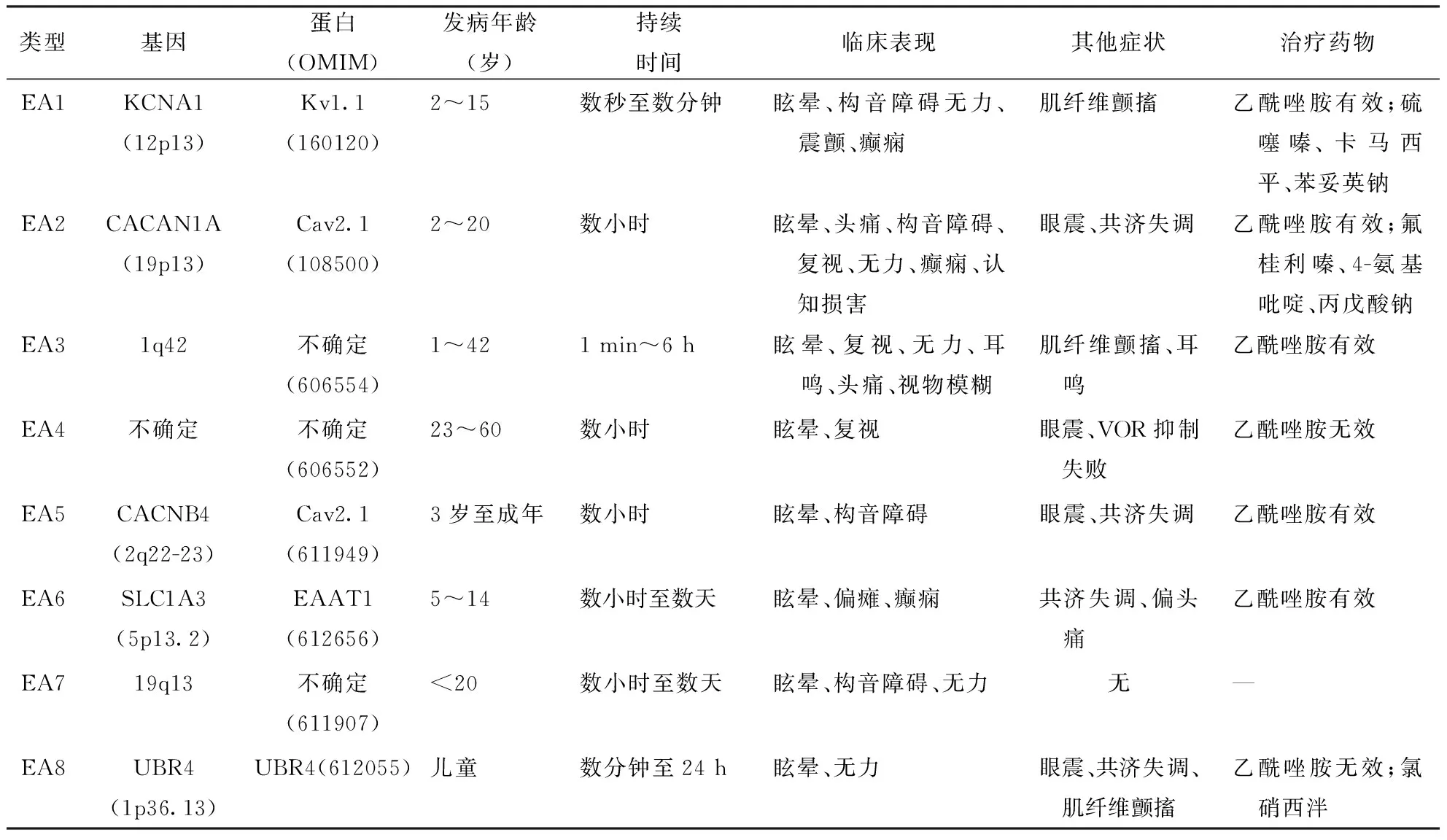
表1 不同类型EA基因、临床表现及治疗用药情况比较
注:8种EA均为常染色体显性遗传;OMIM:在线《人类孟德尔遗传》数据库(Online Mendelian Inheritance in Man)
2.2EA2
2.2.1发病机制:EA2是由编码神经元P/Q型电压依赖性钙离子通道Cav2.1 ɑ1亚单位CACNA1A点突变所致,移码和拼接位点突变可导致翻译过早终止,产生截短和无功能性通道,该突变基因位于染色体19pl3,由47个外显子组成,外显率为80%~90%,其表达在大脑、小脑以及神经肌肉接头,尤其表达在小脑的浦肯野纤维以及颗粒细胞,控制细胞膜的兴奋性和神经递质的释放[12]。EA2发病率低于1∶100 000。另外偏瘫型偏头痛(FHM)、脊髓小脑性共济失调6型(SCA6)也为CACNA1A突变所致,目前发现有60种不同的杂合点突变可导致EA2[13-14]。在少数EA2患者,直接检测CACNA1A序列不能发现任何点突变,但多重链接探针扩增技术可发现大片段的缺失或重复。
2.2.2临床表现:EA2主要表现为发作性共济失调、眼球震颤及构音障碍。患者中50%有偏头痛,某些患者可表现为四肢无力,偶有脑干及皮层受累症状(偏瘫、感觉异常、暗点等);患者中50%出现眩晕、恶心呕吐,约1/3患者眼球震颤多为垂直性且在下视时明显,一般持续数分钟到数天不等,多持续数小时,发作间期症状消失[15],并可见斜颈,智力残疾和精神障碍。发作诱因包括运动、情绪、酒精、苯妥英钠以及咖啡、发热、压力、感染等。EA2婴儿至60岁均可发病,一般症状开始于20岁前,也有少数患者发病年龄较大,发作频率不定,可为1年1~2次,也有1周3~4次者。
2.2.3检查及治疗:EA2患者头颅磁共振可见有小脑萎缩,特别是小脑蚓部萎缩多见。做扫视检查时,EA2患者扫视速度正常,但可见辨距不良[16]。单纤维肌电图可发现颤抖增宽和阻滞。PET检查则显示发作间期整个小脑、丘脑及颞叶前部的糖代谢减低。
生物反馈治疗、避免诱因以及适当锻炼可减少EA2患者发作。乙酰唑胺对于50%~75%的EA2患者有效(起始剂量125 mg/d,直至500~750 mg/d,甚至可以达到1000 mg/d),其效果明显优于EA1[15]。氟桂利嗪和4-氨基吡啶在有些研究中被证实有效。有文献研究报道使用4-氨基吡啶(5 mg 口服 3次/d)对2/3患者可预防复发,对于1/3患者可以减轻症状,其主要作用机制为影响钾离子电流,延长动作电位持续时间,从而达到提高动作电位的传导以及神经递质的释放[17]。该药不良反应比较小,主要为恶心、上腹部不适及精神症状等[17]。丙戊酸钠可以作为辅助治疗用药。
2.3EA3-EA8EA3发现于加拿大一个大家族中,其主要表现为反复发作的眩晕、耳鸣、躯干性共济失调,发作间期肌颤搐、头痛、言语含糊、复视以及无力等,典型患者症状持续1~6 min ,均在30 min之内,发病年龄不等。眩晕、耳鸣以及无眼球震颤、发作时间短有助于EA3与EA1和EA2鉴别。该病基因定位在染色体1q42,和偏头痛性眩晕基因位点重叠,发病年龄较EA1和EA2晚,对于乙酰唑胺治疗有效[18]。
EA4被称为周期性前庭小脑性共济失调,在美国北卡罗来纳有欧洲血统的家系中见诸报道[19]。EA4发病年龄相对较晚,一般在30~60岁发病,主要表现为眩晕、共济失调,以及发作间的眼球震颤,典型症状持续数小时。诱因包括劳累、摇头、视觉刺激等,闭眼可缓解症状。一些患者小脑性共济失调进展缓慢,病变在小脑绒球,前庭眼反射(VOR)抑制功能丧失,不能被乙酰唑胺缓解;EA4基因连锁分析不能归类为EA1和EA2,但是迄今为止没有发现其相关致病基因。
EA5在一些家族中发现,其与CACNB4突变有关,突变位点为染色体2q22-23,致病基因为CACNB4,该基因编码电压门控钙离子通道β4亚单位,此亚单位直接影响Cav2.1的C末端。现有研究发现,在EA5家族中存在C104F错义、R482X无意义突变,同时钾通道功能轻度改变,而且有报告称此基因突变可导致癫痫大发作以及青少年肌阵挛发作。EA5临床表现和EA2类似,主要表现为发作性眩晕和共济失调,可以出现癫痫大发作,一般没有肌纤维颤搐,症状持续数小时,但发病年龄较EA2晚;发作间歇期可见情绪障碍、凝视诱发的眼震、轻度构音障碍和躯干共济失调,对于乙酰唑胺治疗有效[20]。
EA6发现于一例患儿,突变位点为染色体5p13.2,基因为SLC1A3,其编码星形胶质细胞中的谷氨酸运载体(EAAT1),于脑干以及小脑中均有表达,在突触中负责谷氨酸摄取。此突变导致高度保守区蛋白残基改变,相关蛋白功能缺失,目前仅发现P290R和C186S两个突变。EA6临床症状相对较重,表现为发作性以及进行性共济失调,可伴有发作性的偏瘫和癫痫,偏头痛、发热,一般无眼震出现,症状持续数小时到数天,发热可诱发上述症状;EA6患者还可出现视跟踪异常和VOR抑制功能丧失,乙酰唑胺可减少其发作频率[21-22]。
EA7在一四代家族中的7位成员中发现,病变位点为染色体19q13,主要表现为发作性共济失调、无力和构音障碍、眩晕,少数人有偏头痛,症状持续数小时至数天,诱因包括情绪和运动,发作频率为每月1次或数天1次。一般20岁前发病,随着年龄增大,发作频率降低。发病间歇期神经系统查体正常[23]。
EA8发现于一爱尔兰家族中,该家族三代人总共33名患者。病变位点为染色体1p36.13-p34.3。该病多于20岁之前发病,表现为步态不稳、全身无力以及言语含糊,少数患者有眼震、肌纤维颤搐、中度构音障碍、意向性震颤,可有偏头痛,但无癫痫出现,症状持续数分钟至24 h,发病频率高者每日2次,少者数月1次,其中2例女性患者在怀孕期间发作频率减低,诱因为劳累或者压力。余患者均随着年龄增大而发作频率减少。乙酰唑胺治疗无效,但是氯硝西泮有效[3]。
本文报道的患者为青年男性,临床症状严重,以发作性肢体以及躯干共济失调为主要表现,同时为言语含糊,头晕症状较轻,症状每次持续3~4 h,发作时无耳鸣听力下降,无视物旋转、恶心呕吐等。结合患者既往癫痫诊断明确,无脑血管病的危险因素,且MRA以及颈部血管彩超基本正常,脑血管病不考虑;发作时脑电图检查未见异常,故其发作性共济失调不能用癫痫解释;乙酰唑胺治疗有效,该患者无偏头痛病史,结合以上情况临床上考虑为EA,由于经济原因未行基因检测,故仍不能确定分型。
各型EA疾病早期容易被诊断为后循环缺血、重症肌无力、肌无力综合征等多种疾病,目前诊断主要依靠家族史、诱因、临床表现(发作性的运动障碍、眩晕、偏头痛、癫痫等)、查体以及实验室检查(头颅MRI、肌电图、前庭功能检查、基因检测)确定。
综上,EA为一组中枢神经系统离子通道疾病,其具体发作机制尚不清楚。该病虽然不影响患者的生存期,但是严重影响生活质量,所以早期诊断治疗,预后良好。
参考文献:
[1]Jen JC,Graves TD,Hess EJ,et al.Primary episodic ataxias: diagnosis,pathogenesis and treatment[J].Brain,2007,130 (Pt 10): 2484-2493.
[2] Kevin A,Joanna C,Hane Lee,et al.A new episodic ataxia syndrome with linkage to chromosome 19q13[J].Arch Neurol,2007,64(5):749-752.
[3] Conroy J,McGettigan P,Murphy R,at al.A novel locus for episodic ataxia:UBR4 the likely candidate[J].Eur J Hum Genet,2014,22:505-510.
[4] Browne DL,Gancher ST,Nutt JG,et al.Episodic ataxia/myokymia syndrome is associated with point mutations in the human potassium channel gene,KCNA1[J].Nat Genet,1994,8(2):136-140.
[5] Ferrick-Kiddie EA,Rosenthal JJ,Ayers GD,et al.Mutations underlying episodic ataxia type-1 antagonize Kv1.1 RNA editing[J].Sci Rep,2017,7:41095.
[6] D’Adamo MC,Hasan S,Guglielmi L,et al.New insights into the pathogenesis and therapeutics of episodic ataxia type 1[J].Front Cell Neurosci,2015,9:317.
[7] Mestre TA,Manole A,MacDonald H,at al.A novel KCNA1 mutation in a family with episodic ataxia and malignant hyperthermia[J].Neurogenetics,2016,17(4):245-249.
[8] Graves TD,Cha YH,Hahn AF,et al.Episodic ataxia type 1: clinical characterization,quality of life and genotype-phenotype correlation[J].Brain,2014,137(Pt 4):1009-1018.
[9] Tomlinson SE,Tan SV,Kullmann DM,et al.Nerve excitability studies characterize KV1.1 fast potassium channel dysfunction in patients with episodic ataxia type 1[J].Brain,2010,133(Pt/2): 3530-3540.
[10] KotagalV.Acetazolamide-responsive ataxia[J].Semin Neurol,2012,32(5): 533-537.
[11] Eunson LH,Rea R,Zuberi,SM,et al.Clinical,genetic and expression studies of mutations in the potassium channel gene KCNA1 reveal new phenotypic variability[J].Ann Neurol,2000,48(4):647-656.
[12] Mantuano E,Veneziano L,Jodice C,et al.Spinocerebellar ataxia type 6 and episodic ataxia type 2: differences and similarities between two allelic disorders[J].Cytogenet Genome Res,2003,100(1-4):147-153.
[13] Choi KD,Choi JH.Episodic Ataxias:Clinical and Genetic Features[J].J Mov Disord,2016 ,9(3):129-35.
[15] Jen J,Kim GW,Baloh RW.Clinical spectrum of episodic ataxia type 2[J].Neurology,2004,62(1): 17-22.
[16] Baloh RW,Yue Q,Furman JM,et al.Familial episodic ataxia: clinical heterogeneity in four families linked to chromosome 19p[J].Ann Neurol,1997,41(1): 8-16.
[17] Strupp M,Kalla R,Claassen[J],et al.A randomized trial of 4-aminopyridine in EA2 and related familial episodic ataxias[J].Neurology,2011,77(3):269-275.
[18] Cader MZ,Steckley JL,Dyment DA,et al.A genome-wide screen and linkage mapping for a large pedigree with episodic ataxia[J].Neurology,2005,65(1):156-158.
[19] Damji KF,Allingham RR,Pollock SC,et al.Periodic vestibulocerebellar ataxia,an autosomal dominant ataxia with defective smooth pursuit,is genetically distinct from other autosomal dominant ataxias[J].Arch Neurol,1996,53(4):338-344.
[20] Escayg A,De Waard M,Lee DD,et al.Coding and noncoding variation of the human calcium-channel beta4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia[J].Am J Hum Genet,2000,66(5):1531-1539.
[21] Merrill MJ,Nai D,Ghosh P,et al.Neuropathology in a case of episodic ataxia type 4[J].Neuropathol Appl Neurobiol,2016,42(3): 296-300.
[22] Jen JC,Wan J,Palos TP,et al.Mutation in the glutamate transporter EAAT1 causes episodic ataxia,hemiplegia,and seizures[J].Neurology,2005,65(4):529-534.
[23] Kerber KA,Jen JC,Lee H,at al.A new episodic ataxia syndrome with linkage to chromosome 19q13[J].Arch Neurol,2007,64(5):749-752.
猜你喜欢
中国医药导报(2022年28期)2022-11-25
中华养生保健(2020年2期)2020-11-16
中国医学影像技术(2020年11期)2020-01-13
创新作文(小学版)(2019年4期)2019-07-24
女士(2017年10期)2017-11-01
饮食科学(2017年5期)2017-05-20
哲思2.0(2017年12期)2017-03-13
安徽医科大学学报(2015年9期)2015-12-16
恋爱婚姻家庭·养生版(2015年12期)2015-05-14
为了孩子(孕0~3岁)(2001年3期)2001-06-13
