
肉芽肿性多血管炎合并IgG4相关性肾病
2018-05-05 02:41梁丹丹曾彩虹
肾脏病与透析肾移植杂志 2018年2期
梁丹丹 曾彩虹
病例摘要
病史患者男性,55岁,因“消瘦、乏力2个月,血清肌(SCr)酐升高1月”于2017-12-28入院。
患者近2月感明显消瘦、乏力,体重减轻20 kg。1月前至外院查尿常规:尿隐血+,尿蛋白-,尿微量蛋白/肌酐57 mg/g,血红蛋白(Hb) 79 g/L,SCr 224.5 μmol/L,血清白蛋白(Alb) 26.5 g/L,肝功能、血脂正常,未予相关治疗。2周前至我院门诊查尿蛋白定量0.88 g/24h,尿红细胞-,Hb 81 g/L,Alb 34.90 g/L,SCr 287.3 μmol/L,为进一步检查及治疗入院。病程中患者无皮疹、关节痛、腹痛等,目前精神尚可,食欲、睡眠、大便及排尿正常。
既往史20年前发现“高血压病”,未定期监测血压。1年前诊断“2型糖尿病”,自行口服降糖药物,未监测血糖。3月前发现“两侧脑室旁及两侧基底节区腔梗”。否认过敏史。
家族史父母均有“高血压病”,否认家族中同样疾病患者。个人史无特殊。
体格检查体温37℃,脉搏90次/min,呼吸18次/min,血压152/90 mmHg,体质量指数22.5 kg/m2;神清,咽不红,扁桃体不大,心、肺、腹部未见明显异常,双下肢无水肿。
实验室检查
血常规 Hb 67 g/L,红细胞2.49×1012/L,白细胞8.1×109/L,血小板250×109/L,嗜酸性粒细胞7.2%,网织红细胞3.35%。
尿液 尿蛋白定量1.05 g/24h,尿沉渣红细胞计数4.3/μl;尿C3 2.0 mg/L(正常值≤2.76 mg/L),α2-MG 2.0 mg/L(正常值≤2.87 mg/L),NAG 21.1 U/(g·Cr)[正常值≤16.5 U/(g·Cr)],RBP 7.8 mg/L(正常值<0.5 mg/L),Lyso<0.5 mg/L。
血生化 Alb 34.4 g/L,球蛋白18.0 g/L,尿素氮24.1 mmol/L,SCr 260.8 μmol/L,尿酸383 μmol/L,谷丙转氨酶34 U/L,谷草转氨酶17 U/L,总胆固醇2.94 mmol/L,三酰甘油0.97 mmol/L,钠136.3 mmol/L,钾3.08 mmol/L,氯96.2 mmol/L,总二氧化碳25 mmol/L,钙2.12 mmol/L,磷1.23 mmol/L。
其他 ANA 1∶ 256,ENA抗体谱、A-dsDNA阴性。IgG 17.6 g/L,IgA 2.33 g/L,IgM 0.577 g/L,IgE 54.2 IU/ml,补体C3 1.15 g/L,C4 0.269 g/L,ASO、RF正常。血免疫球蛋白亚类(IgG4)定量测定4,710(正常范围30~2 010)mg/L。MPO-ANCA 42.53 RU/ml。血管紧张素转换酶21.0 U/L(正常范围0~52 U/L)。抗结核抗体和结核感染T细胞检测阴性。血免疫固定电泳未见单克隆条带。血游离κ轻链/λ轻链比值正常,β2微球蛋白(血)7.32 mg/L、铁蛋白622.1 μg/L、其他肿瘤标志物阴性。红细胞碎片和溶血检查均阴性。HbA1c 5.5%。传染病四项均阴性。骨髓细胞学检查大致正常。

图1 A:肾间质增宽、水肿,大量炎细胞浸润,以单个核细胞为主,亦见较多浆细胞、中性粒细胞,见肉芽肿形成(↑)(HE,×200);B:肾间质见较多浆细胞聚集分布(HE,×400);C:肾间质见“席纹”状纤维化(PASM-Masson,×400);D:肾小球毛细血管袢皱缩、开放欠佳,囊壁见分层,周围间质大量炎细胞浸润(PAS,×400);E:肾间质血管壁破坏,管壁见纤维素样物质沉积(PASM-Masson,×400);F:肾间质血管Fibrin阳性(IF,×200)
影像学检查
超声 双肾B超:左肾120 mm×55 mm×60 mm;右肾120 mm×65 mm×60 mm,右肾轻度积水,双肾皮质回声稍增强。腹部超声:(1)胆囊息肉;(2)胰腺未见肿大;(3)肝脾声像图所见范围未见占位。(4)门静脉彩色多普勒检查未见异常。心脏超声示轻度二尖瓣反流,LVEF:70%。
CT 头颈部CT示:(1)左侧上颌窦、右侧蝶窦小囊肿;(2)双侧下鼻甲肥大,鼻中隔偏曲。颈胸部CT示:(1)右肺中叶结节灶,考虑纤维硬结;(2)左肺下叶钙化灶;右肺下叶及左肺散在少许炎症;(3)主动脉及冠状动脉粥样硬化。腹部CT示:(1)左肾上腺弥漫性稍增粗,考虑增生可能;(2)盆腔积液,腹壁软组织水肿。
心电图 (1)窦性心律;(2)qⅢ、aVF≥1/4r3、T波改变。
肾活检病理
光镜 皮质肾组织3条。肾小管间质重度急性病变伴慢性病变,多灶性肾小管上皮细胞扁平、刷状缘脱落,多灶性肾小管萎缩、基膜增厚,部分肾小管损毁,数量减少,间质弥漫增宽、水肿,纤维化+~++,偶见“席纹”状纤维化,大量炎细胞浸润,以单个核细胞为主,亦见大量浆细胞、少量嗜酸性粒细胞及中性粒细胞,间质中可见数处肉芽肿样组织,部分中心见纤维素样坏死,有的肉芽肿形成于坏死的血管周围,有的包绕于废弃肾小球周围(图1A~C)。31个肾小球中6个球性废弃。余肾小球毛细血管袢皱缩、开放欠佳(图1D),系膜区未见明显增宽,囊壁增厚、分层,伴囊周纤维化,个别球囊壁破坏。PASM-Masson:阴性。数处间质血管坏死、结构破坏(图1E),周围大量炎细胞浸润,数处小动脉管壁增厚、闭锁,一处小动脉内皮细胞增生、肿胀,管腔狭窄。
免疫荧光 冰冻切片未见肾小球。石蜡切片常规荧光染色、轻链染色均阴性。Fibrin染色示数处血管阳性(图1F)。
电镜 观察2个肾小球。肾小球系膜区偶见增宽,系膜区未见电子致密物分布。肾小球毛细血管袢基膜略皱缩,开放欠佳,节段袢内皮下疏松、增宽,基膜厚约330~620 nm。肾小球毛细血管袢基膜内皮下、上皮侧均未见电子致密物分布。肾小球足细胞足突节段融合(30%~40%),胞质少量微绒毛化,胞浆内见吞噬性溶酶体。间质大量炎细胞浸润,见单个核细胞、浆细胞、中性粒细胞等,邻近见肾小管炎,肾小管基膜未见电子致密物分布。
皮肤脂肪组织刚果红染色未见阳性。荧光染色示κ轻链、λ轻链均为阴性。
免疫组化肾组织IgG4染色示肾间质CD138+平均细胞数目为532个/mm2(图2A),IgG4+细胞为50个/HP(图2B)。
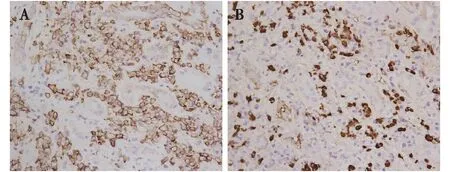
图2 A:肾间质见较多CD138+浸润细胞;B:肾间质IgG4+细胞>50个/HP(IH,×400)
小结:肉芽肿性多血管炎合并IgG4相关性肾病。
讨 论
患者中年男性,起病急,肾脏损害表现为急性肾功能不全,中少量蛋白尿,伴高血压、贫血,无血尿、水肿等,肾外症状有消瘦、乏力、体重下降。血MPO-ANCA阳性,血IgG、IgG4水平升高。肾脏B超提示双肾肿大。肾活检病理示急性间质性肾炎、间质血管炎症性坏死及坏死性肉芽肿形成,而肾小球仅表现为毛细血管袢缺血皱缩,无明显增生、坏死性病变,免疫组化提示肾间质IgG4+细胞为50个/HP。
该患者突出的病理表现之一为间质数处动脉壁坏死、结构破坏、周围伴炎细胞浸润,即动脉炎症性坏死性改变,伴间质坏死性肉芽肿形成。根据2012年Chapel Hill会议标准,血管炎分为大、中、小血管炎。其中大血管炎包括大动脉炎和巨细胞动脉炎,主要累及主动脉及其分支,而该患者大血管并未见明确病变,病变部位为肾小叶间动脉。中血管炎包括结节性多动脉炎和川崎病,川崎病主要发生于婴幼儿,多累及冠状动脉,结节性多动脉炎则重点需与ANCA相关性血管炎进行鉴别,而ANCA抗体是鉴别两者的主要标志物。小血管炎则根据血管壁是否有免疫复合物沉积分为免疫复合物性血管炎和寡免疫复合物性血管炎。该患者临床未见系统性红斑狼疮、类风湿性关节炎、感染、恶性肿瘤等继发性病因相关的阳性体征和检查结果,且肾组织(包括肾小球及血管)免疫荧光常规及轻链染色均阴性,抗肾小球基膜肾病、过敏性紫癜性肾炎及冷球蛋白血症相关血管炎等可能性不大,补体水平正常,低补体血症性荨麻疹性血管炎亦排除,因此考虑寡免疫复合物性血管炎,即ANCA相关性血管炎。其分为显微镜下多血管炎、肉芽肿性多血管炎和嗜酸性肉芽肿性多血管炎三种。该患者MPO-ANCA阳性,肾组织除间质血管炎外,尚见间质多处坏死性肉芽肿形成,患者临床无哮喘症状,血嗜酸性粒细胞、IgE比例不高,最终考虑为肉芽肿性多血管炎。针对患者肾间质中的非干酪样坏死性肉芽肿,还需鉴别结节病、肾结核等,而相关检查提示血清血管紧张素转换酶水平正常、结核阴性,因此均予以排除。
该患者突出的病理表现之二为急性间质性肾炎。间质性肾炎的病因复杂,包括药物、感染、肿瘤、自身免疫性疾病、代谢性疾病等多方面,其中大部分继发病因通过临床表现和实验室检查已排除,淋巴瘤则通过免疫组化染色已排除。对于该患者,值得注意的是肾组织浸润细胞除大量单个核细胞外,亦见多灶性聚集的浆细胞,因此需考虑IgG4相关性疾病的可能。IgG4相关性疾病是一个系统性疾病,表现为多脏器炎症伴纤维化和硬化,伴或不伴血清中IgG4水平升高。组织学改变表现为大量淋巴细胞和浆细胞浸润,伴轻-中度嗜酸性粒细胞浸润,“席纹”状纤维化,闭塞性静脉炎,而中性粒细胞浸润和肉芽肿形成均罕见。淋巴细胞由T细胞和B细胞构成,其中B细胞可以形成生发中心,而T细胞则弥漫分布在病变部位。浆细胞的四种IgG亚型均可见,并以IgG4为主。如果腺体受累,这些浸润细胞易聚集在导管周围[1-4]。这些炎症性病变可引起相应脏器的肿胀,破坏正常结构。肾脏受累主要表现为间质性肾炎,少数可见肾小球病变和浆细胞动脉炎,动脉炎一般无动脉壁坏死和弹性纤维断裂。目前的诊断标准是肾组织免疫组化染色显示IgG4+细胞>10个/HP或IgG4+细胞/IgG+细胞比值>40%提示IgG4相关性肾病可能性大[5]。该患者临床表现为肾脏肿大,血清IgG、IgG4水平升高,肾组织学提示间质大量浆细胞浸润伴少量嗜酸性粒细胞,其中IgG4+细胞更高达50个/HP,亦见特征性的“席纹”状纤维化,因此支持IgG4相关性肾病的诊断。IgG4相关的间质性肾炎可导致临床出现少量蛋白尿、肾功能下降及贫血等多种实验室检查改变。此外,既往亦有文献报道ANCA阳性患者也可表现为间质性肾炎[6-8],而该患者血MPO-ANCA阳性,尚不能完全明确其是否同时造成肾间质损害。
组织IgG4+浆细胞增多和血清IgG4水平升高并非IgG4相关性疾病所特有。其他多种疾病可导致组织IgG4+浆细胞增多和血清IgG4水平升高,包括ANCA相关性血管炎、多中心型Castleman病、恶性肿瘤、干燥综合征、炎症性肠病、类风湿性关节炎和原发性硬化性胆管炎等[9-11]。其中ANCA相关性血管炎与IgG4相关性疾病的很多临床表现和组织学特征存在交叉和相似,按照2011年的专家共识,诊断IgG4相关性疾病时需排除ANCA相关性血管炎[5],但是目前对这点是存在疑问的。Chang等[12]对46例肉芽肿性多血管炎患者和20例对照进行组织IgG和IgG4免疫组化染色,结果显示4例鼻窦组织和4例眶内/眶周组织IgG4+细胞37~139个/HP,IgG4+/IgG+细胞比值44%~83%。很多其他研究也发现部分ANCA相关性血管炎(尤其肉芽肿性多血管炎和嗜酸性肉芽肿性多血管炎)的组织活检标本可见较多IgG4+细胞浸润,部分IgG4相关性疾病患者可伴血清ANCA阳性,似乎两者容易共存[13-16]。目前,ANCA相关性血管炎和IgG4相关性肾病发病的病理生理学机制均未完全清楚,多项研究证实嗜酸性肉芽肿性多血管炎患者不仅血IgG4水平升高,且与疾病的活动情况有关[17-18],这也提示两者的发病机制可能相近或存在某种关联。有研究表明两种疾病中滤泡辅助性T细胞(Tfh)均增加,且以Tfh2细胞为主,而Tfh2细胞会导致IgG4+浆细胞的水平增加[19-20],进一步完善两者发病机制的研究可能会提高对这种疾病状态的认识。就目前来看,鉴别是否IgG4相关性疾病主要还是结合临床表现和影像学检查,组织学检查中席纹状纤维化、闭塞性静脉炎是其相对较特异的病理改变。
肉芽肿性多血管炎和IgG4相关性疾病均为系统性疾病,除肾脏受累外,亦可累及全身其他多个脏器。该患者肺部CT示纤维结节,尽管临床无咯血等症状,仍高度考虑与这两种疾病存在关联,因未行肺活检,不能明确其性质及确切病因。对其他系统的筛查暂未发现受累证据,比如头颈部CT未发现ANCA相关性血管炎相关的鼻窦病变,腹部CT亦未见胰腺肿大。这两种疾病均需要免疫抑制剂治疗,该患者诊断明确后给予甲泼尼龙冲击,后续口服泼尼松联合霉酚酸酯治疗1月,目前SCr降至138.8 μmol/L,尿蛋白转阴,血清MPO-ANCA及IgG4水平均较入院时下降。
小结:IgG4相关性疾病是一种少见的系统性疾病,与ANCA相关性血管炎的临床表现和组织学改变存在一些相似和交叉。ANCA相关性血管炎经典的肾脏病理改变为坏死性新月体肾炎、血管炎,可伴肉芽肿形成,部分病例可表现为间质性肾炎,部分病例可出现间质IgG4阳性细胞浸润,此时需结合临床与IgG4相关性疾病进行鉴别。当前对两者的认识仍存在局限性,进一步研究两者的发病机制有助于阐明两者的联系。
1 Deshpande V,Gupta R,Sainani N,et al.Subclassification of autoimmune pancreatitis:a histologic classification with clinical significance.Am J Surg Pathol,2011,35(1):26-35.
2 Zen Y,Nakanuma Y.IgG4-related disease:a cross-sectional study of 114 cases.Am J Surg Pathol,2010,34(12):1812-1819.
3 Zen Y,Inoue D,Kitao A,et al.IgG4-related lung and pleural disease:a clinicopathologic study of 21 cases.Am J Surg Pathol,2009,33(12):1886-1893.
4 Kawa S,Okazaki K,Kamisawa T,et al.Japanese consensus guidelines for management of autoimmune pancreatitis:II.Extrapancreatic lesions,differential diagnosis.J Gastroenterol,2010,45(4):355-369.
5 Kawano M,Saeki T,Nakashima H,et al.Proposal for diagnostic criteria for IgG4-related kidney disease.Clin Exp Nephrol,2011,15(5):615-626.
6 Schönermarck U,Schirren CA,Mistry-Burchardi N,et al.Interstitial nephritis and high titers of PR3-ANCA:an unusual manifestation of ANCA-associated disease.Clin Nephrol,2005,64(5):383-386.
7 Wen YK,Chen ML.Transformation from tubulointerstitial nephritis to crescentic glomerulonephritis:an unusual presentation of ANCA-associated renal vasculitis.Ren Fail,2006,28(2):189-191.
8 Nakabayashi K,Sumiishi A,Sano K,et al.Tubulointerstitial nephritis without glomerular lesions in three patients with myeloperoxidase-ANCA-associated vasculitis.Clin Exp Nephrol,2009,13(6):605-613.
9 Yamamoto M,Tabeya T,Naishiro Y,et al.Value of serum IgG4 in the diagnosis of IgG4-related disease and in differentiation from rheumatic diseases and other diseases.Mod Rheumatol,2012,22(3):419-425.
10 Ebbo M,Grados A,Bernit E,et al.Pathologies Associated with Serum IgG4 Elevation.Int J Rheumatol,2012,2012:602809.
11 Ryu JH,Horie R,Sekiguchi H,et al.Spectrum of Disorders Associated with Elevated Serum IgG4 Levels Encountered in Clinical Practice.Int J Rheumatol,2012,2012:232960.
12 Chang SY,Keogh KA,Lewis JE,et al.IgG4-positive plasma cells in granulomatosis with polyangiitis (Wegener′s):a clinicopathologic and immunohistochemical study on 43 granulomatosis with polyangiitis and 20 control cases.Hum Pathol,2013,44(11):2432-2437.
13 Ayuzawa N,Ubara Y,Keiichi S,et al.Churg-Strauss syndrome with a clinical condition similar to IgG4-related kidney disease:a case report.Intern Med,2012,51(10):1233-1238.
14 Perez AR,Martínez C,Espinoza LR.IgG4-associated vasculitis.Curr Rheumatol Rep,2013,15(8):348.
15 Della-Torre E,Lanzillotta M,Campochiaro C,et al.Antineutrophil cytoplasmic antibody positivity in IgG4-related disease:A case report and review of the literature.Medicine (Baltimore),2016,95(34):e4633.
16 Danlos FX,Rossi GM,Blockmans D,et al.Antineutrophil cytoplasmic antibody-associated vasculitides and IgG4-related disease:A new overlap syndrome.Autoimmun Rev,2017,16(10):1036-1043.
17 Vaglio A,Strehl JD,Manger B,et al.IgG4 immune response in Churg-Strauss syndrome.Ann Rheum Dis,2012,71(3):390-393.
18 Yamamoto M,Takahashi H,Suzuki C,et al.Analysis of serum IgG subclasses in Churg-Strauss syndrome--the meaning of elevated serum levels of IgG4.Intern Med,2010,49(14):1365-1370.
19 Zhao Y,Lutalo PM,Thomas JE,et al.Circulating T follicular helper cell and regulatory T cell frequencies are influenced by B cell depletion in patients with granulomatosis with polyangiitis.Rheumatology (Oxford),2014,53(4):621-630.
20 Akiyama M,Suzuki K,Yamaoka K,et al.Number of Circulating Follicular Helper 2 T Cells Correlates With IgG4 and Interleukin-4 Levels and Plasmablast Numbers in IgG4-Related Disease.Arthritis Rheumatol,2015,67(9):2476-2481.
猜你喜欢
开卷有益·求医问药(2022年8期)2022-09-29
现代医院(2022年6期)2022-08-26
中国临床医学(2022年3期)2022-07-08
天津医科大学学报(2021年3期)2021-07-21
复旦学报(医学版)(2021年2期)2021-04-09
西南国防医药(2016年7期)2016-12-01
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
中华老年多器官疾病杂志(2016年8期)2016-05-14
中华皮肤科杂志(2014年4期)2014-12-19
