
Gaussian 09软件在配合物紫外-可见吸收光谱教学中的应用
2018-10-18 07:16吴杰颖
赤峰学院学报·自然科学版 2018年9期
张 琼,吴杰颖
(安徽大学 化学化工学院,安徽 合肥 230601)
Gaussian软件包用于执行各种半经验和从头分子轨道计算,是目前应用最广泛的计算化学软件之一[1],Gaussian 09软件是Gaussian系列计算软件的最新版本,其主要功能可用来预测分子在真空或者溶液状态下的优化结构、电子排布、紫外-可见吸收光谱、红外和拉曼光谱、核磁谱、分子极性等性质.Gaussian View是Gaussian软件的图形用户界面,它主要的用途有两个方面:(1)构建输入文件;(2)直观显示计算结果.对于要用Gaussian软件研究的体系,首先要在Gaussian View中构建分子结构,保存为.gjf文件,然后输入Gaussian程序中计算,在构建的过程中,需要注意键长、键角、空间构型和对称性等方面的准确性.此外,Gaussian View与多种图形软件兼容,可以读入Chem3D和晶体数据等多种格式文件,大大拓宽了它的使用范围.
金属配合物的分子轨道是由金属的价轨道和配体对称性匹配的群轨道线性组合而成,涉及成键、反键、非键等多种轨道类型.最低激发态的电子构型受配位方式影响.图1为过渡金属配合物(八面体构型)的分子轨道能级示意图[2],金属到配体的电荷转移(MLCT)是电荷从金属d轨道跃迁到配体π*轨道,配体到金属的电荷转移(LMCT)是电荷从配体转移到金属.其他跃迁方式包括配体到配体电荷跃迁(LLCT)、配体内电荷跃迁(ILCT)及金属中心(MC)电荷跃迁.
电子的跃迁在紫外-可见吸收光谱的教学过程中较抽象,很难靠传统的教学方法获得良好的教学效果.将Gaussian软件应用于配合物的紫外-可见吸收光谱教学中,具体做法是:以课题组合成的多种过渡金属配合物为研究对象,利用配合物的紫外-可见吸收光谱的实验结果[3-4],在单晶结构基础上,采用含时密度泛函理论(TD-DFT)计算配合物的跃迁.结构优化采用未进行对称限制的B3LYP泛函进行,然后在优化结构的基础上采用B3LYP泛函进行TD-DFT计算.结构优化及TD-DFT计算均采用Gaussian 09程序进行.对于基态的优化及最低的25个单线态到单线态的激发能量的TD-DFT计算采用6-31G*基组 (对于C、H、N、O 原子)及 LanL2dz 基组(对于 Pt、Zn、Cl、Br原子).理论结合实验,可以帮助学生更形象地理解金属配合物的电子跃迁方式,收到较好的教学效果.

图1 八面体构型配合物的分子轨道能级示意图
1 配体内的电荷跃迁

图2 (a)TDPt结构 (b)由实验获得配体TSD及配合物TDPt的紫外-可见吸收光谱(c-d) 通过Gaussian计算获得TDPt的前线分子轨道电子云分布图
配体的π轨道中的电子受激发跃迁到反馈π*空轨道(π→π*)称为配体内的电荷跃迁(intraligand charge transfer,ILCT),ILCT对应的紫外-可见吸收特征峰一般在紫外区.ILCT具有强吸收,ε值在104数量级,金属中心对ILCT干扰小,紫外-可见吸收特征峰几乎不受其他辅助配体影响.
紫外-可见吸收光谱结果表明:配合物在310nm处的紫外可见吸收光谱ε值为5×104mol-1L cm-1,仅比配体在305nm处的吸收峰红移5nm,可归属于ILCT.理论计算结果表明:在配合物分子HOMO-1轨道中,电子云主要分布在三苯胺官能团,在LUMO+2轨道中,电子云分布在N,N二乙基苯胺官能团,电子云从HOMO-1轨道跃迁至LUMO+2轨道,可明显观察到ILCT,通过轨道间的能级差ΔE计算出的紫外-可见吸收光谱特征峰的位置与实验结果吻合,为解释电子跃迁提供了有力的证明.
2 金属到配体的电荷跃迁
金属中心d轨道到有机配体π*轨道的跃迁称为金属到配体的电荷跃迁 (metal-to-ligand charge transfer MLCT),金属中心d轨道和配体π*轨道较低能级间隙导致紫外-可见吸收特征峰出现在长波区.MLCT通常包含容易氧化的金属中心和具有低能空轨道的配体.MLCT具强吸收,ε值在104数量级或更大,金属中心和配体会影响MLCT特征峰,调节配体可调控峰的位置.
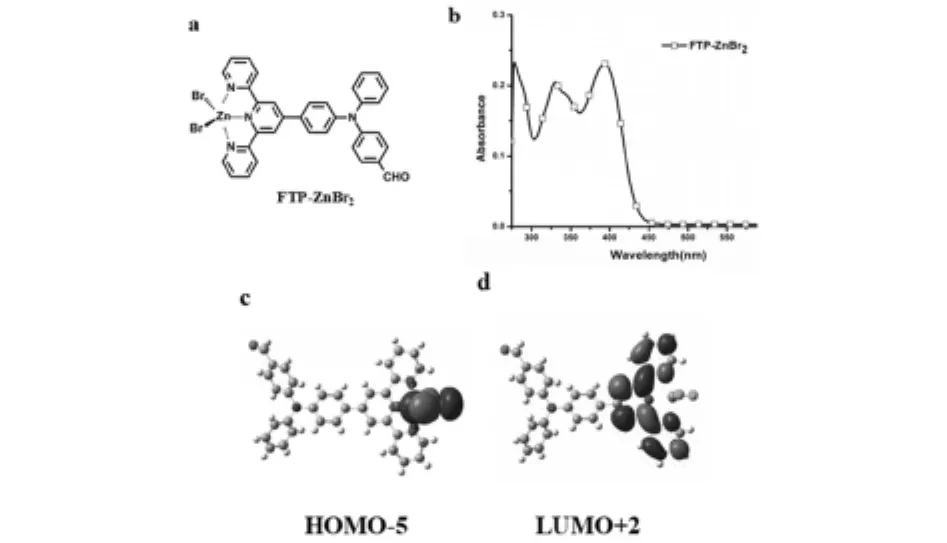
图3 (a)FTP-ZnBr2结构 (b)由实验获得FTP-ZnBr2的紫外-可见吸收光谱 (c-d)通过Gaussian计算获得FTP-ZnBr2的前线分子轨道电子云分布图
紫外-可见吸收光谱结果表明:配合物在405nm处的紫外-可见吸收光谱ε值为2.5×104mol-1L cm-1,可归属于MLCT.理论计算结果表明:在配合物分子的HOMO-5轨道中,电子云主要分布在中心金属原子(Zn),在LUMO+2轨道中,电子云分布在三吡啶官能团,可以明显地观察到MLCT,通过HOMO-5与LUMO+2的能级差ΔE计算出的紫外-可见吸收光谱特征峰为410nm,与实验值基本一致.
3 配体到配体的电荷跃迁
当配合物中含有两种或多种配体时,其中一种配体最高占有轨道能级高于金属离子的最高占有d轨道,产生配体到配体的电荷跃迁 (Ligand-to-ligand charge transfer LLCT).LLCT的吸收一般属中等强度吸收,ε值一般在103~104数量级.LLCT跃迁受配体的取代基影响较大,不同的供吸电子基团可使紫外-可见吸收特征峰发生红移/蓝移.
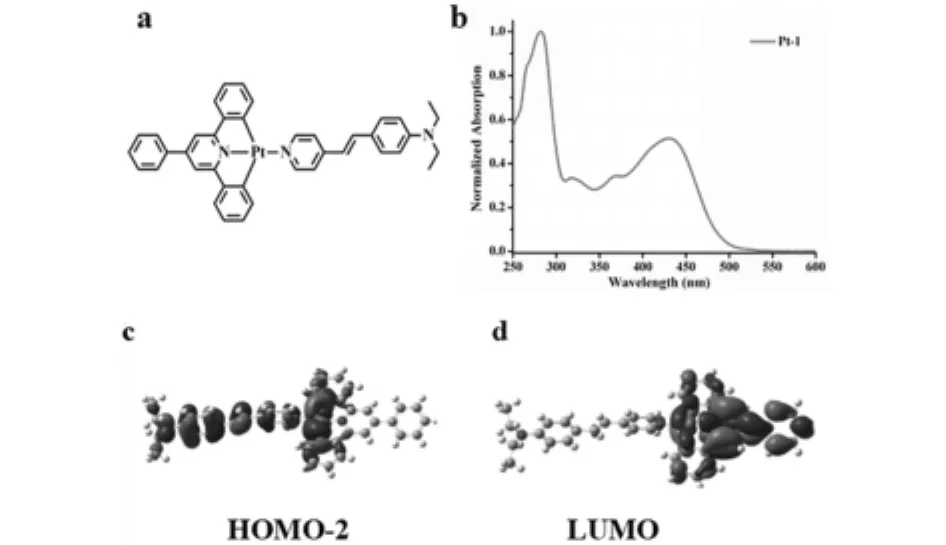
图4 (a)Pt-1结构 (b)由实验获得Pt-1的紫外-可见吸收光谱 (c-d)通过Gaussian计算获得Pt-1的前线分子轨道电子云分布图
紫外-可见吸收光谱表明:Pt-1在320nm和375nm处的吸收峰的ε值大约为4×104mol-1L cm-1,可以归属为LLCT.理论计算结果表明:在Pt-1的HOMO-2轨道中,分子的电子云主要分布在N,N-二乙基苯乙烯基吡啶上.在LUMO轨道中,分子的电子云主要分布在6'-苯基-2,2'-联吡啶上,电子从HOMO-2轨道跃迁至LUMO轨道,即LLCT,通过轨道间能级差ΔE计算紫外-可见特征峰位置与实验结果相吻合.
综上所述,在配合物的紫外-可见吸收光谱教学中,通过Gaussian软件的引入能够在教学过程中对于配合物中的电子激发态给学生提供更为形象的描述,通过理论计算,可以让学生掌握电子云分布的抽象概念,也可以让学生验证实验结果,极大地激发学生学习兴趣,提高教学质量和教学效果.
猜你喜欢
中学生数理化·中考版(2021年10期)2021-11-22
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中国资源综合利用(2017年1期)2018-01-22
材料科学与工程学报(2016年4期)2017-01-15
山东工业技术(2016年15期)2016-12-01
中国粮油学报(2016年5期)2016-01-23
合成化学(2015年4期)2016-01-17
新高考·高一物理(2015年6期)2015-09-28
新高考·高一物理(2015年6期)2015-09-28
郑州大学学报(理学版)(2014年3期)2014-03-01
