
铯在钨(110)表面吸附行为的第一性原理研究
2019-02-14 01:27郑剑平钟武烨
原子能科学技术 2019年1期
丁 硕,郑剑平,钟武烨
(中国原子能科学研究院 反应堆工程技术研究部,北京 102413)
钨是一种具有较高表面功函数的难熔金属,以(110)表面为例,其表面功函数高达5.0~5.2 eV[1-4],而未经特定表面处理的多晶钨的功函数也可达4.5 eV左右[1-3,5-12]。根据理论分析和实验结果,具有较高功函数的金属表面通常会表现出较低的铯吸附表面功函数[13]。因此,铯原子吸附的钨(110)表面通常用作高性能的电子发射材料,广泛应用于热离子能量转换器[14-18]、光电转换器[19]、离子推进系统[20]及用于高效加热聚变等离子体的中性束注入装置的离子源[21]等。铯原子在钨表面的吸附位置和吸附率可通过低能电子衍射(LEED)和俄歇电子光谱技术(AES)[4]确定。多项研究[3-5,10]表明,铯在钨表面的吸附功函数随铯原子吸附率的变化有规律地呈U形曲线。随铯原子吸附率的增加,表面功函数先降低后缓慢增加,在约2.1 eV达到稳定,其中,在铯原子吸附率约为0.25时表面功函数达到最小值1.4~1.6 eV,该数值低于纯铯(110)表面的功函数,说明铯原子在钨表面的吸附效应造成了表面功函数的降低。
就表面功函数的测量而言,由于铯原子极强的腐蚀性和化学活性,通过现有的双电极单元或开尔文探针原子力显微镜对特定铯压下具有特定铯原子吸附率的表面系统进行安全而可靠的测量是较为困难的。因此,通过第一性原理方法对铯吸附系统的表面功函数进行高精度的理论计算可避免进行有铯存在时危险而难以控制的实验,同时可得到表面吸附结构和特定晶面上更精确的功函数。本文通过基于密度泛函理论的第一性原理计算方法,研究铯原子在钨(110)表面上的吸附行为。
1 计算方法
本文计算过程均是通过Materials Studio 8.0软件中的密度泛函理论方法进行的。原子的吸附位置通过Adsorption Locator模块中的模拟退火方法确定,而结果优化和系统特性的计算则通过CASTEP模块完成[22-24]。基于密度泛函理论,计算中采用平面波超软赝势,并使用BFGS算法对晶体结构进行弛豫。采用的赝势外层电子结构配置为:W,5s25p65d46s2;Cs,5s25p66s1。选择GGA PW91交换关联泛函进行计算。所有的计算均在虚空间中进行,且在Brillouin区积分中采用Monkhorst-Pack划分并加入特殊高对称k点。
为防止采用实验晶格参数导致的人工应力和虚假弛豫[24],对BCC钨的晶格参数进行高精度收敛计算。对于1×1×1钨晶胞,截断能取600 eV,k点数为16×16×16,并对长程力采用默认的OBS修正[25]。收敛参数设置为:能量变化<5×10-7eV/atom,外力<107eV/m,应力<0.002 GPa,且位移变化<5×10-15m。计算得到的晶格常数为3.169×10-10m,与实验值3.165×10-10m符合较好。本工作所有模型均是依据该计算得到的晶格参数建立的,以便与计算设置相匹配。

a——BCC钨的晶体结构;b——偶极子修正后的钨(110)2×2表面模型;c——钨(110) 1×1表面模型图1 计算所用的原子结构Fig.1 Atomic structure for calculation
图1为计算所用的原子结构,其中被几何约束和未进行几何约束的原子分别用红色和灰色球体表示,叠层模型可用于表面吸附计算。通过计算得到的表面功函数与实验值的收敛测试可知,对于钨(110)表面可使用5层原子的叠层模型,其中,顶部2层原子处于自由状态,并对下部3层原子的绝对坐标和相对坐标进行几何约束。计算中采用自适应偶极子修正方法[26]以便对叠层模型中产生的偶极子进行补偿。由于原子叠层的厚度约为8.96×10-10m,真空层的厚度设置为4×10-9m以确保在相邻的叠层之间几乎无相互作用,从而保证表面功函数计算值的精确度。
计算中选用模拟退火法确定铯原子在钨(110)表面上的最大吸附率和粗略的吸附位置。选用12×12和24×24超晶格的表面作为计算基体以避免边界效应对计算结果的影响。计算的循环次数设定为≥20,且每个循环的步数≥106。为提高吸附计算的效率,铯吸附计算的空间被限制在通过原子集确定的表面附近的空间内部,约是表面原子层上方厚度为10-9m的范围内。对于12×12和24×24表面,向系统内添加的铯原子数分别为300和600个,从而确保吸附原子足够过量以使表面第1吸附层被填满。
对于吸附后的晶胞而言,几何优化计算的截断能设定为350 eV,且对于钨1×1晶胞其k点数≥10×10×1,为确保得到理想结果,收敛参数设置为:能量变化<5×10-6eV/atom,外力<10-8eV/m,应力<0.02 GPa,且位移变化<5×10-14m。
2 结果与讨论
2.1 最大吸附率
钨表面铯原子的吸附率η的计算公式为:
η=nads/nsurf
(1)
其中:nads为吸附原子数;nsurf为吸附基体的第1层原子数。
由于铯原子的半径远大于钨原子的半径,根据体积效应,铯原子的最大吸附率应<1.0。钨(110)表面12×12和24×24超晶格采用Adsorption Locator的计算结果如图2所示,铯原子、被几何约束和未进行几何约束的钨原子分别用紫色、红色和灰色球体表示。其中,被单层铯原子覆盖的满吸附钨表面的结构形式如图2b、c所示。

a——钨(110)表面12×12的原始结果;b——钨(110)表面12×12和24×24的第1吸附层;c——钨(110)表面12×12和24×24的钨基体表层图2 采用Adsorption Locator的计算结果Fig.2 Result calculated by Adsorption Locator
在钨(110)表面12×12和24×24的吸附系统中,第1吸附层和钨基体表层的原子数和吸附率列于表1,可看出,单层铯原子在钨(110)表面的最大吸附率约为0.4,而文献[4]的实验结果为0.5,产生不同的原因可能是对钨表面铯原子具体吸附模式的认识不同,也可能是实验中的表面情况不理想。
2.2 吸附位置
吸附原子在基体表面常见的吸附位置包括:顶位、三原子中心、长桥和短桥,如图3所示。根据能量最低原则,通常采用吸附能Eads作为确定吸附原子实际吸附位置的依据:
Eads=Etotal-Eslab-nCsECs
(2)
其中:Etotal、Eslab、ECs分别为吸附系统总能量、吸附基体能量和铯原子能量;nCs为吸附系统中的铯原子数。
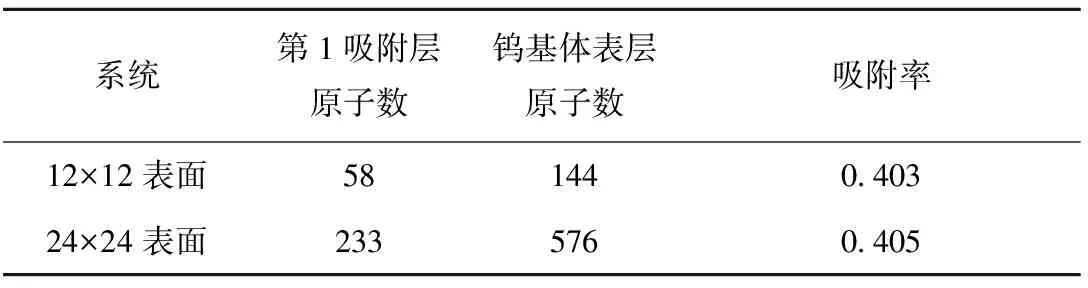
表1 第1吸附层和钨基体表层原子数和吸附率Table 1 Atom number of the first adsorption layer and top tungsten surface layer and adsorption rate

图3 钨(110)表面上不同的吸附位置Fig.3 Different adsorption locations on W(110) surface
铯原子在钨表面的实际吸附位置随吸附率的变化而变化。当铯原子吸附率较低时,如吸附率为0.25时,将铯原子分别置于顶位、三原子中心、长桥和短桥上方,并对铯原子x和y方向的位移进行约束。当几何优化过程结束后,最终的原子结构和对应的吸附能列于表2。根据吸附能的计算结果,低吸附率下最可能的吸附位置是长桥。
值得注意的是,当铯原子吸附率较高,甚至在基体表面形成完整的吸附单原子层时,文献[4]提出了铯原子占据不同的三原子中心位置形成具有0.5吸附率的平面密排结构单原子层。然而,该结构并非平面密排结构,不同位置的铯原子之间的距离差异较大(超过10-10m),无法形成平面六边形密排结构。事实上,当吸附原子形成平面密排结构时,铯原子实际占据了两种能量较低的位置,包括短桥和三原子中心。此时,铯原子沿密排方向按照-ABA′B′-结构排列,如图4所示,其中A和A′代表不同的三原子中心序列,B和B′代表不同的短桥序列,整体表面吸附率为0.4。该现象发生的原理是,当吸附率较高时,铯原子的体积效应成为决定性的因素,铯原子无法占据低吸附率下能量最低的长桥位置,而是被迫吸附在不同的能量次低位置,从而形成平面密排结构以保证整个系统的能量最低。

表2 不同吸附系统的吸附能Table 2 Adsorption energy of different adsorption systems
2.3 功函数
对于热电子发射材料来说,表面功函数是其最重要的性质之一。功函数ψ是真空能级Evac和费米能级EFermit的差值,可通过第一性原理计算方法得到:
ψ=Evac-EFermit
(3)
根据Richardson-Dashman方程,热电子发射电流密度J与功函数ψ的关系为:
(4)
其中:A为与表面特性有关的常数;T为温度;k为Boltzmann常数。
对已进行自适应偶极子修正的具有不同表面吸附率的表面模型进行电势分析,可得到足够精确的功函数。为进行对比,建立6层原子的铯(110)叠层模型以进行纯铯(110)表面的功函数计算。铯原子层的厚度(17.3×10-10m)和真空层的厚度(8×10-9m)按照钨吸附模型的比例选取。
不同吸附率下的表面功函数如图5所示,铯吸附功函数随铯原子吸附率的变化呈U形曲线变化,且计算得到的功函数和文献[4]的实验结果符合较好。

a——三原子中心铯吸附结构;b——-ABA′B′-铯吸附结构;c——24×24吸附表面的密排方向和由黑线划定的周期型结构;d——吸附结构晶格的俯视图;e——吸附结构晶格的透视图图4 铯吸附率为0.4时的吸附位置Fig.4 Adsorption location with cesium adsorption rate of 0.4

图5 不同吸附率下的表面功函数Fig.5 Surface work function of different adsorption rates
当铯原子吸附率约为0.25时,吸附系统的表面功函数小于纯铯(110)的表面功函数,该现象说明钨表面的吸附效应影响了表面铯原子的状态,而这一效应可能是通过改变表面电特性和铯原子外层电子能量造成的。假设在铯原子在钨(110)表面的吸附系统中,铯原子和钨基体原子无相互作用,铯原子和钨基体原子各自独立发生热电子发射现象,则当铯原子未达到满吸附时,吸附表面的整体功函数应是铯原子吸附率的线性函数,等于铯原子和钨基体表面功函数依照表面原子数的加权平均;而当铯原子达到满吸附后,吸附系统表面函数即为铯原子的功函数(图6)。

图6 铯原子和钨基体不存在相互作用时系统吸附功函数随铯原子吸附率的变化Fig.6 Adsorption work function change with cesium atom adsorption rate without interaction between cesium atom and W(110) surface
由于铯原子吸附在钨(110)表面时两者存在相互作用,如吸附表面存在电场重分布作用和吸附原子的电子能量状态重分布效应,吸附系统功函数的实验值则呈现与上述线性函数变化规律不同的特性。
2.4 偶极子模型和分态密度
偶极子模型[26]常用于解释吸附表面的功函数变化。根据该模型,在吸附过程中,铯原子的外层电子向基体表面转移,使得铯原子本身发生极化或电离现象。转移的电子与钨基体中的自由电子结合在表面附近共同形成偶极子结构,进而改变表面电势的分布,并使功函数减小。图7为电子密度差计算结果。可知,铯原子吸附后,钨基体表面附近电子密度增加的区域扩大,电子密度减小的区域缩小,表明铯原子向基体表面发生了电子转移。
随吸附率增大,吸附的铯原子电子总量增多,去极化效应变得显著,这意味着当极化效应和去极化效应达到平衡时,表面功函数达到最小值。此后,偶极子结构继续发生去极化效应,最后吸附表面功函数与纯铯表面功函数相同。电子能量的分布可通过能带积分,即态密度表示。分态密度的计算结果则可用来评估不同电子轨道在态密度中的贡献。铯原子吸附率为0.25时铯原子与纯铯表面铯原子的态密度计算结果如图8所示。

a——铯吸附系统;b——纯铯(110)表面图7 电子密度差计算结果Fig.7 Calculation result of electron density difference

图8 态密度计算结果Fig.8 Calculation result of DOS
费米能级附近的能带对功函数的特性具有决定性的影响,吸附态铯原子的能带范围为-10.75~0 eV,而纯铯中铯原子的能带范围为-11.36~0 eV。从图8可看出,吸附态铯原子能带的分布范围明显较纯铯的小,说明吸附效应使铯原子的电子发生了局域化效应。与纯铯表面相比,吸附对电子能量分布的局域化效应降低了铯原子真空能级和费米能级之间的差距,从而降低了表面功函数(图9)。
3 结论
通过基于密度泛函理论的第一性原理计算方法研究了铯原子在钨(110)表面的吸附行为。Adsorption Locater模块的计算结果表明,铯在钨(110)表面的单原子层最大吸附率为0.4,铯原子的吸附位置随铯原子吸附率的增大而变化,当铯原子吸附率为0.25时铯原子优先占据长桥位置,当铯原子吸附率为0.4时铯原子以-ABA′B′-结构吸附在钨基体表面。随铯原子吸附率的增大,表面功函数先减小后增大,最终稳定在2.134 eV,其中,功函数的最小值1.524 eV出现在吸附率为0.25时。该最小值低于纯铯(110)表面的功函数。功函数减小的机理可通过偶极子模型和分态密度来解释。在偶极子模型中,从吸附态铯原子向钨基体表面的电子转移机制是表面功函数降低的原因,而分态密度计算结果表明吸附过程对铯原子电子能量分布的影响对表面功函数的降低亦有贡献。

a,c——态密度;b,d——s轨道分态密度图9 态密度和分态密度计算结果Fig.9 Calculation result of DOS and PDOS
猜你喜欢
石材(2022年3期)2022-06-01
石材(2022年3期)2022-06-01
湖南大学学报(自然科学版)(2021年8期)2021-09-27
化工管理(2021年7期)2021-05-13
海洋学报(2021年1期)2021-03-02
中国材料进展(2019年5期)2019-07-20
成都信息工程大学学报(2019年5期)2019-05-21
小学生学习指导(高年级)(2017年10期)2017-09-15
小天使·三年级语数英综合(2017年6期)2017-06-07
橡胶工业(2015年8期)2015-07-29
