
Geoscience material structures prediction via CALYPSO methodology
2019-11-06 00:45AndreasHermann
Chinese Physics B 2019年10期
Andreas Hermann
Centre for Science at Extreme Conditions and SUPA,School of Physics and Astronomy,The University of Edinburgh,Edinburgh,EH9 3FD,United Kingdom
Keywords:crystal structure prediction,core materials,planetary ices,hydrous minerals
1.Introduction
Understanding the interior makeup of and the dynamical processes within the planets in our and other solar systems is a central research focus in geo-and planetary sciences.Quantifying planets’bulk chemical composition is essential to distinguishing and validating models of planetary formation and thermal evolution over the lifetime of the solar system.More detailed pictures of stratification inside planets are necessary to understand planets’ability to harbour life–from establishing stable planetary magnetic fields by dynamos of convecting conducting material to sustaining surface water by plate tectonics and deep water storage.At the heart of these phenomena are the properties of materials(elastic,conductive,etc.)under high-pressure and-temperature conditions as found inside planetary bodies.These states of matter are mostly solid(about 85%of Earth’s volume,70%of its mass,are in the solid state)and they are not easily accessible by direct observation or laboratory experiments. Accurate calculations are therefore indispensable to assist or guide experimental efforts.Crystal structure prediction(CSP)has emerged as a very versatile and useful tool in surveying properties of materials under high-pressure conditions,in particular if combined with accurate density functional theory(DFT)calculations.[1]Materials systems relevant to geosciences come with their own challenges,these are discussed in more detail in the next section. However,CSP also offers unique opportunities to the field.It successfully challenges conventional chemical beliefs by uncovering new paradigms about reactivity that run counter to the chemical intuition trained at ambient conditions.It can uncover important phase transitions that in turn lead to new phase relations with severe geochemical implications.And it provides starting points(through the most relevant solid structures)for additional computational studies,e.g.,molecular dynamics,that can explore high-temperature phenomena.
In this brief review,we aim to discuss some the successes of CSP,in particular from the CALYPSO code,[2]in various areas of geoscientific interest.We begin by outlining in Section 2 some of the unique challenges faced by CSP if applied to the geoscience context.In Section 3 we discuss the application of CSP to understand planetary core materials,via the incorporation of volatile elements in iron-rich solid cores.In Section 4 we discuss CSP applications to the mantle regions of icy planets,and in Section 5 the mantle regions of rocky planets.Section 6 offers a brief summary and outlook.
2.Methodological challenges
CSP faces some rather unique challenges if applied to geologically relevant scenarios. There are over 5000 classified minerals.[3]This might not seem terribly many:databases hold over 500 unique allotropes of carbon alone,[4]over 200000 inorganic(ICSD[5])and over 1000000 organic and metal-organic structures(CSD[6]). However,minerals come with an unusual chemical complexity: many minerals contain four or more different elements(minerals with over 20 elements have been reported[7]). This increases the dimensionality of the crystalline configuration space,which is the bottleneck for an efficient application of CSP.The minerals we know are exclusively from our studies of Earth’s crust and mantle and from meteorites.It is very likely that other environments(e.g.,icy worlds,rocky planets with different chemical composition,or even the lower depths of Earth)will support a wide range of other mineral materials. One potential avenue to restrict the computational cost of structure searches in such complicated settings is to exploit common structural features of minerals such as regular polyhedral coordination:silicon usually appears at the center of SiO4tetrahedra or(at high pressures)SiO6octahedra.Constructing structural candidates from such reasonable building blocks instead of individual atoms would reduce the number of irrelevant structures considerably;of course the CSP code needs to support such features and correctly interpret their presence when deducing the next generation of candidates;and one must ensure that such“chemical coarsening”does not restrict the searches at the conditions in question.This process can be automated[8]and combined with machine-learning(ML)techniques to train computationally cheap interaction potentials[9,10]to explore much larger and more complex structures than hitherto possible.
As a second key property,materials in geological environments invariably encounter high temperatures. CSP usually produces ground state results,applying the‘clamped-nuclei’approximation to obtain the electronic and Ewald energies of the solid state structures. At finite temperature,entropy can play a decisive role in stabilising certain materials:this could be configurational entropy due to atomic disorder,or vibrational entropy due to thermal occupation of lattice vibrations,i.e.phonons. The first case is very relevant for minerals,which often form as solid solutions. For example,alloying of magnesium and iron atoms is common because the ionic radii of Mg2+and Fe2+are very similar. Materials that are stabilised by the resulting configurational entropy are hard to capture with the default CSP approach that uses the enthalpy,H=E+pV,as fitness function to rank(and promote)certain structures.The second case,vibrational entropy,can in principle be captured by performing phonon calculations on candidate structures,but this would increase the computational cost of the structure searching process by 1–2 orders of magnitude.The current approach is to assume strong overlaps between the sets of structures with lowest free energies and lowest enthalpies;such that the latter can be used to determine vibrational entropies a posteriori for the most relevant candidate structures only.Of course,atomic interaction potentials e.g.constructed by ML techniques would be a promising addition to CSP in this case as well:if the potentials are accurate enough to capture atomic forces they could provide a computationally affordable alternative to derive vibrational free energies.
Despite these challenges,CSP is beginning to make an impact in the field of geosciencess,as the case studies in the following sections will illustrate.
3.Volatiles in iron-rich cores
Earth’s solid inner core is dominated by an iron-rich Fe-Ni alloy[11,12]but its overall density is significantly lower than expected from such an alloy. The core must therefore contain some light elements,dissolved at the single per cent level.The main candidates,from chemical and geochemical arguments,are H,C,O,Si,P,and S.[13,14]Most of these elements form one or more stoichiometric compounds with iron and/or nickel;[15]these have been studied extensively by experiment and ab initio calculations.CSP arguably has less of a role to play for these(often quite simple)compounds,yet it has meaningfully contributed to the study of several binary Fe–X phase diagrams.We will discuss these in turn below.
Fe–O.Iron oxides are notoriously difficult to describe in DFT,due to the correlated nature of the Fe-3d electrons that are responsible for magnetism and a Mott insulating state,e.g.in FeO.Under pressure,however,the magnetism collapses and iron oxides undergo insulator-metal transitions;the resulting high-pressure phases are much more straightforward to capture with standard DFT,and therefore accessible to CSP approaches. The most remarkable recent result from such searches is the prediction of a new iron oxide,FeO2,to become stable at conditions relevant at the bottom of Earth’s mantle and in Earth’s core.[16–18]It forms in the cubic pyrite structure type,with FeO6octahedra strongly tilted such that they develop O–O“bonds”to adjacent octahedra(see Fig.1).The character of these“bonds”,and their influence on the oxidation state of the iron atoms,have been cause for some debate:if they signify the presence of elongated anionic oxygen molecules,(O2)2−,then iron has the common Fe2+oxidation state(FeO2would be a peroxide),whereas if they do not represent any chemical bonding(the O–O separation is much longer than in(O2)2−)iron would have a highly unusual Fe4+oxidation state.The truth is likely somewhere in the middle.[18,19]The hydrous analogue of the new oxide,FeOOHx(at ambient pressure x=1 is known as goethite,and commonly as rust),would alleviate the concerns over the oxidation state,could act as a major water reservoir at the core-mantle boundary(CMB)and is a candidate material to form the“ultra-low velocity zones”present at the CMB.However,there is controversy over whether FeOOH would retain or release hydrogen at CMB conditions.[20]A subsequent CSP study found FeOOH to remain stable and undergo further phase transitions at pressure conditions close to the inner core.[21]FeOOH essentially retains the pyrite-type structure of FeO2,with hydrogens inserted into the long O–O bonds present in FeO2(see Fig.1).Hence,in both compounds the Fe atoms form cornersharing FeO6polyhedra.Moreover,FeO2was found to react with helium to form FeO2He at pressures above 120 GPa.[22]The latter compound differs structurally(if not electronically)from the others.In contrast to FeO2and FeOOH,iron atoms in FeO2He-Fmm form corner-sharing FeO8polyhedra,with helium occupying the interstitial cubic sites.This compound is isostructural to full Heusler compounds X2YZ;moreover,it is isostructural to another high-pressure helium compound,Na2He,[23]if the latter is interpreted as an electride,with a localised electron pair occupying a lattice site.

Fig.1.Crystal structures of iron oxide FeO2 and derived compounds.(a) at 76 GPa;[17](b) at 100 GPa;[21](c)FeO2He at 100 GPa.[22]Fe(O,H,He)atoms are gold(red,pink,white)spheres,respectively.
Fe–H.The formation of iron hydride FeH under pressure has been achieved several decades ago[24]and in recent years polyhydrides FeH3[25](above 86 GPa)and FeH5[26](above 120 GPa)have been reported. Early DFT calculations focused on known simple structure types[27]but CSP has since contributed meaningfully in exploring the binary Fe–H phase diagram,[28–31]both motivating and confirming the recent experimental efforts.The resurgent interest in iron hydrides in recent years is due to the promise of high-Tcsuperconductivity in compressed metal hydrides,[32]but in this case has consequences for our understanding of the makeup of Earth’s core as well.Clearly the pressure conditions inside the core support formation of iron polyhydrides;it is less clear whether they would remain solid at core temperatures.In any case,the hydrogen-poor environment of the core would point towards formation of a solid solution of FeH and iron over the polyhydrides.
Fe–C.Two relatively simple iron carbides are known and have been subjected to detailed experimental and DFT investigations: Fe3C,an iron-rich compound important in metallurgy,[33]and Fe7C3, which forms at relatively low pressures.[34,35]Searching beyond these known compounds,CSP has been used by two groups to find other candidates for high-pressure Fe–C compounds.[28,36]Both reported a new phase,Fe2C,to become stable at inner core pressure conditions,which has not been seen in experiments so far.Meanwhile,a very recent CSP study suggested a new high-pressure structure for Fe7C3that is more stable than the known structure at pressure conditions in the inner core.[37]Figure 2 compares the crystal structures of these compounds:both Fe3C and Fe7C3feature carbon atoms in hexagonal Fe6prisms;these CFe6prisms are edge-and corner-sharing in both Fe3C and Fe7C3,with higher degree of connectivity in the latter.Fe2C on the other hand has eight-fold coordinated carbon,inside quite irregular Fe8polyhedra that fill more of space than in the more iron-rich compounds.

Fig.2.Crystal structures of iron carbides.(a)Fe3C-Pnma,(b)Fe7C3-P63mc,(c)Fe2C-Pnma,all at 300 GPa and drawn to the same scale.[28]Fe(C)atoms are gold(black)spheres.
Fe–SiandFe–S.Both silicon and sulfur form very simple compounds with iron.Iron silicide FeSi has an intriguing structure at ambient pressure[38]and undergoes a phase transition to the CsCl structure type that was predicted[39]before seen in experiment.[40]Subsequent CSP efforts have essentially confirmed this structural sequence and so far failed to find other stable iron silicides at high pressure conditions.[41]Iron sulfide FeS has a rich phase diagram,and the highest pressure phase solved in experiments[42]has subsequently been confirmed in a CSP study,which also suggested a new phase stable at yet higher pressures.[43]
(Fe,Ni)–Xe.While xenon is clearly not one of the volatiles to make up a sizeable portion of Earth’s core,its incorporation in minerals is nonetheless of fundamental interest. The“missing xenon”problem refers to the depleted abundance of xenon in Earth’s atmosphere relative to interstellar abundance ratios of the elements.[44,45]One potential explanation is that xenon can be bound in mineral form deep inside Earth.Increased reactivity of the noble gas elements under pressure has been seen in numerous CSP studies[46–50]and for the purpose of this section,xenon was found to react with either iron or nickel at inner core pressure and temperature conditions.[46]Xenon is predicted to form XeM3compounds with either metal,which are in parts stabilised by charge transfer from xenon to iron or nickel.The total amount of charge transferred is not very large(about 0.24/0.21 electrons per Fe/Ni atom)but is nonetheless chemically intriguing,as examples of anionic Fe/Ni are rare. Several other XeMnstoichiometries were also reported to become stable at high pressures while,notably,neither krypton nor argon were found to form stable compounds. This distinction for xenon hints at its special ability to form compounds at planetary pressure conditions;however,other mineral routes for the“missing xenon”need to be accounted for,such as incorporation in silicates[51,52]or as trace element in the liquid outer core;the latter would provide a viable route to retain xenon inside the early Earth before potentially precipitating in the solid core.
While both experiments and calculations have made great strides in studies of volatile elements interacting with iron,there are many more challenges ahead before a full understanding of the composition of heavy planetary cores can emerge.Other light elements could be present in the core,with magnesium a prime candidate;more studies are needed on Mg–Fe mixtures,which might tilt the balance of whether magnesium can partition in iron instead of silicates.[15]The ternary phase diagrams Fe–Ni–X for some volatile element X might look different than both the binary phase diagrams Fe–X or Ni–X,and this would further influence the volatiles’propensity to form compounds in a core of Earth’s composition.[53]And super-Earth exoplanets might require thinking about much more extreme pressure conditions in their cores.[54]These are formidable challenges but CSP is well poised to help answer these questions.
4.Icy mantles
The interiors of giant icy planets,such as Uranus and Neptune in our solar system,are altogether quite different environments. Their mantle regions comprise mixtures of molecular ices of water,ammonia and methane,together with impurities and volatiles such as hydrogen or helium. Similar mixtures are presumed to occur prominently in the large number of Neptune-like exoplanets discovered by recent and ongoing astronomical observation campaigns.[55–59]
Little is known how the molecular ingredients arrange themselves within these planetary bodies.They could segregate to form layers of distinct compositions,or homogeneous mixtures at roughly the overall chemical composition ratio.
4.1.Individual molecules

Fig.3.High pressure water ice P21 at 2 TPa,interpreted in different ways:(a)as(H3O)+(OH)−visualized by ELF=0.75 isosurface,[60](b)as hcplike packing of O atoms,visualized by quasi-six fold symmetry,[61](c)as high-coordination phase beyond tetrahedral connectivity.[62](d)Pressure evolution of O–H separations in the structure.[62]
CSP has tremendously expanded our knowledge about individual ices’response to high-pressure conditions. For water,the cubic phase ice-X was for decades the highest pressure phase known[63]before a DFT molecular dynamics study in 1996 suggested a transition of ice-X to an orthorhombic Pbcm phase around 300 GPa.[64]Phases beyond the Pbcm phase were successfully constructed manually in 2010,[65]but immediately following that work a series of papers explored water into the TPa pressure range using CSP and converged on a very rich phase diagram that features layered structures,metallic phases,and scope for decomposition of water into H2O2and H-intercalated water phases.[60–62,66,67]This series of CSP studies allows for interesting insight into this type of research:even though several groups found a specific high-pressure phase of ice,of P21symmetry,their interpretations differ significantly(see Fig.3):where one group saw partial ionisation(the formation of(H3O)+(OH)−),[60]another saw hexagonal close packing of oxygen,slightly distorted by the presence of protons,[61]and yet another saw the emergence of higher coordination,the deviation from tetrahedral connectivity so dominant in ice phases.[62]Even though CSP gave the same structure,our understanding of the underlying physics and chemistry can depend a lot on the researchers’point of view;it is vital to have these different view points and discussions to ultimately form a better picture of the driving forces behind phase transformations and changes in material properties under pressure.The lessons thus learned will be invaluable not just for geosciences but also materials science and solid state chemistry.The ground state phase evolution for water that was established in this way has since formed the foundation for other computational studies,e.g.of water’s high-temperature properties and interesting states such as diffusive protons.[68,69]These states are now explored in state-of-the-art dynamic compression experiments.[70,71]
Methane,CH4,follows a very different route under pressure,and our understanding of its P-T phase diagram arguably suffers much more from significant disagreements between theory and experiment. It is likely that a lot of these disagreements are related to the metastability inherent to hydrocarbon chemistry,but also to failures of ab initio methods to survey free energy landscapes accurately. The seminal contribution of CSP to our understanding of methane was made in 2010,[72]when CALYPSO was used to search for highpressure phases of CH4.A sequence of phase transitions of methane into longer hydrocarbons culminated in the full decomposition into diamond and hydrogen at just below 300 GPa at low temperatures,which should happen much earlier at elevated temperatures.Such a decomposition(or any reaction of methane to form longer hydrocarbons)was not seen in static room temperature compression experiments–either using optical absorption measurements,[73]x-ray diffraction[74]or Raman spectroscopy.[75]However,it was reported that higher temperatures induced polymerisation of methane at around 1100 K,eventually leading to diamond formation at 3000 K and very low pressures of just 10 GPa.[76,77]Since the initial CSP report on methane,a sequence of other works have explored the hydrocarbon(C–H)phase diagram.[78–80]A recent revisit of the phase diagrams of methane and other hydrocarbons assembled all these results for the first time,augmented by additional CSP runs,and pointed to the prominence of molecular van der Waals compounds(CH4)m(H2)n,which are expected to extend the stability regime of CH4molecules to higher pressures.[81]As a consequence,a full binary phase diagram of C–H compounds can be drawn up,yielding stable hydrocarbon phases as function of pressure,temperature,and composition,see Fig.4.Such a broader approach towards hydrocarbon chemistry is needed as it better connects to experimental efforts to explore planetary interior conditions by dynamic compression of polystyrene,polyethylene,and similar plastic materials.[82–84]

Fig.4.Phase stability chart of hydrocarbon compounds CHn at T=500 K from the harmonic approximation.Coloured horizontal lines indicate stability of respective phases,with metastability(less than 10 meV/atom above the convex hull)indicated by thin black lines.Taken from Ref.[81].
Ammonia,NH3,reacts differently again under pressure.In another success for CSP,a sequence of phase transitions to ionic ammonium amide(NH4)+(NH2)−phases was first predicted[85]and subsequently confirmed in experiment.[86,87]A broader study of hydronitrogens predicts the decomposition of NH3into NH3and N3H7above 440 GPa.[88]
A minor component of icy planets is hydrogen sulfide,H2S.A CSP exploration of its high-pressure properties rightly focused on its metallisation and high-Tcsuperconductivity.[89]Its role within icy planetary bodies,in particular through interactions with other molecular species,could prove quite important,but has not been explored in much detail.
In that context an important question is whether properties of mixtures of these molecular ices can be obtained by averaging over their individual properties–the so-called linear mixing approximation has seen significant successes based on individual molecules’equations of state[90,91]–or whether molecular mixtures can features unique chemistry that needs to be considered in planetary models.
4.2.Molecular mixtures
The low-pressure phase relations amongst the ices of water,methane,and ammonia can be summarised in the ternary phase diagram shown in Fig.5(a).Methane and water form a sequence of clathrate hydrate compounds at low pressures;these have been discovered in experiments and comprise complex water‘host’networks with‘cages’that can hold methane‘guest’species.[92]There is some uncertainty about the filling of the‘cages’and therefore the overall stoichiometry of these hydrates.Remarkably,water and methane have been reported to exhibit significant miscibility(up to 40%methane content)in the fluid state at 2 GPa.[93]Ammonia and water are different as they can form hydrogen-bonded networks,which are arguably less complex in structure.Three compositions of ammonia hydrates have been explored around ambient and low-pressure conditions(up to around 10 GPa):ammonia monohydrate(AMH,NH3:H2O=1:1),ammonia dihydrate(ADH,1:2)and ammonia hemihydrate(AHH,2:1).[94,95]All three hydrates feature internal phase transitions,with five solid AMH and ADH phases,as well as three solid AHH phases identified in experiment.For reference,the solar abundance ratio of the three molecules is also included in Fig.5(a).

Fig.5. (a)Ternary phase diagram of H2O,NH3 and CH4,with known methane hydrates phases(“MH-I”etc),ammonia hydrates(“ADH”etc.),and the solar abundance ratio(grey circle)indicated.(b)ELF=0.7 isosurface of ammonia quarterhydrate(AQH)at 100 GPa,outlining spherical oxygen anions O2−and cations.O(N,H)atoms are red(blue,pink)spheres,respectively.Dashed lines indicate hydrogen bonds.
There is currently no CSP study of this full ternary molecular composition space.However,CALYPSO has proved invaluable e.g.in exploring mixtures of ammonia and water under pressure.[96,97]These recent studies reported sequences of new high-pressure phases for the three canonical mixing ratios,as well as the stabilisation of a new ammonia-rich hydrate of 4:1 stoichiometry(ammonia quarter hydrate,AQH).The pervasive feature of the high-pressure phases of these mixtures is proton transfer from water to ammonia.Depending on stoichiometry this results in ammonium hydroxidesammonium oxidesand other ionic speciessee Fig.5(b)for an example. As a consequence,high pressure is found to stabilise ammonia-rich hydrates that benefit most from the ionic bonding enabled by this charge transfer,which is opposite to the solar abundances of water and ammonia.The wealth of ground state structural data on ammonia hydrates should now be used to explore their finite temperature properties.This might lead to different conclusions than previous computational studies[98,99]that were in parts based on crystal structures now shown to be metastable.
Molecular mixtures should not only comprise the ices introduced above but also their interactions with the lighter compounds,namely hydrogen and helium,which are known to form outer atmospheres of large icy planets. These light species can potentially interact with the molecular ices and lead to interesting interactions at the boundary of the planets’atmospheres and mantles.Molecular hydrogen is particularly interesting in that regard.We know hydrogen can interact with water,as it forms a sequence of molecular hydrogen hydrates under pressure[92]–and new hydrates with novel water networks are still being reported.[100–102]There is a miscibility gap of H2and H2O(at least in the solid state)beyond the molecular hydrate phases[103]and a re-emergence of atomic hydrogen interacting with the layered network phases of water at TPa pressures.[67]Hydrogen can also interact with methane to form van der Waals compounds that feature prominently in the predicted phase diagrams of hydrocarbons.[81]These compounds include CH4(H2)2,which has one of the highest releasable hydrogen contents by weight(20%)of any material;albeit at very high pressures.However,arguably the chemically most interesting case is hydrogen mixing with ammonia:a recent CALYPSO CSP study[104]found that NH3(H2)2is stabilised at relatively low pressures by formation of ammonium,which develops strong ionic and hydrogen bonds to atomic anionic hydrogen,H−.Its releasable hydrogen content is 19 wt-%. As in the case of ammonia hydrates,the propensity of ammonia to acquire additional protons leads to chemically very interesting structures;and as in the ammonia hydrates,the unusual cationcan form under specific conditions. Finite temperature studies revealed the presence of a superionic regime in this compound.[104]This NH7compound extends knowledge of the binary N–H phase diagram gained in an earlier CSP study.[88]

Fig.6.Molecular mixtures of hydrogen with other molecules,as predicted from CSP.(a)Hydrogen hydrate C3,H2O(H2)2,at 40 GPa;(b)methane hydride CH4(H2)2 at 100 GPa;(c)ammonia hydride NH3(H2)2 at 100 GPa.H(O,C,N)atoms are denoted by pink(red,brown,blue)spheres.All structures are drawn to same scale and have ELF=0.95 isosurfaces.Dashed lines are hydrogen bonds.
Figure 6 gives an overview of these molecular hydrides,with their chemical bonding visualised through the electron localization function(ELF).This showcases their different character:hydrogen in water arranges itself around water’s hydrogen bond network;hydrogen and methane form purely van der Waals bounded complexes;while hydrogen and ammonia are stabilised by the break-up of H2molecules,and unusual hydrogen bond networks emerge of the type
Helium can also form mixtures with small molecules.Most prominently, perhaps, its interaction with water at low pressures results in the formation of clathrate hydrates and filled ices.[105,106]Several recent CSP studies reported helium–water mixtures at much higher pressures and found a variety of stable compounds with stoichiometries across 2:1,1:1,and 1:2.[107–109]These compounds are still dominated by tetrahedral water networks,with their natural cavities occupied by various amounts of helium atoms,see Fig.7. By all accounts the helium does not interact chemically with the water“host”network. Despite the evidence that these compounds are stable,there seems scope for a more comprehensive study since the published data covers disjointed pressure regimes:below 100 GPa[108]and above 300 GPa.[107,109]Similarly,the potential of helium to mix with ammonia has been studied for 1:1 mixtures,[109]but other compositions(likely to form hydrogen-bonded ammonia or ammonium networks)or helium–methane mixtures(likely to be dominated by van der Waals compounds)remain to be explored.

Fig.7. Helium–water mixtures under pressure. (a)He2H2O at 70 GPa,[108](b)He(H2O)2 at 300 GPa.[107]O(H,He)atoms are shown as red(pink,white)spheres,with O–H bonds indicated.
At this point in time,CSP studies of compressed planetary ices and their mixtures far outpace the experimental data. In parts this is testament to the difficulty in performing accurate measurements on these systems under pressure–even constraining mixing ratios in sample chambers is very challenging. Nonetheless,there is much left to do for theory:having unearthed a large variety of new compounds and new chemistry in binary systems,will the same hold true for more complex mixtures?And what can be learned if minority species(such as H2S)are considered as part of the mixtures?
5.Rocky mantles
The mantle regions of rocky planets differ substantially from the scenarios discussed so far.For once,they are at much less extreme pressure and temperature conditions than planetary cores or the mantles of giant icy planets.On Earth,pressures in the upper mantle reach 13 GPa(at 410 km depth),up to 21 GPa at the mantle transition zone(at 660 km depth),and about 125 GPa at the core-mantle boundary(2900 km depth).The minerals that occupy this region are often dominated by ionic bonding and electronically inert,i.e.,wide-gap insulators,which aids calculations.On the other hand,the mantle region is by far the most chemically diverse place of all scenarios discussed in this paper;in fact it is the most chemically diverse region we know in the universe,though that might be biased towards what we can observe.This leads to complicated phase relations between minerals and a plethora of possible chemical reactions,resulting in formation or dissolution of specific compounds.Moreover,many minerals can form solid solutions along continuous chemical substitutions.The simplest example is probably ferropericlase or magnesiowüstite,(Mg,Fe)O,but the same principle also applies to olivine,(Mg,Fe)2SiO4,and garnets,X3Y2Si3O12with X=Ca,Mg,Fe,Mn and Y=Al,Fe,Cr.While in these cases di-and trivalent atoms are exchanged like-for-like,atomic substitutions in minerals can be more complicated;for example,the exchangeis possible. The resulting intermediate members of such solid solutions are stabilised in parts by configurational entropy,which is hard to capture using CSP.Nonetheless,recent years have seen some progress being made in this area.
5.1.Hydrous minerals
The processes by which volatile components such as water,carbon,or nitrogen are cycled between the surface layers and the mantle region of Earth are important to understand Earth’s surface environment.[110–112]Water in particular plays an important role in sustaining and mediating geological activity.The presence of water helps in lowering the mantle’s melting temperature,enhances diffusion and creep,thus affecting rheology of rocks,and also influences mineral phase boundaries.[113]It is estimated that the Earth’s mantle contains a mass of water equivalent to the mass of the world’s oceans.[114,115]At relatively low pressures and temperatures in Earth’s crust,in cold subduction zones,water can exist in molecular form,e.g.in clays such as kaolinite.[116]At highpressure and high-temperature conditions of the mantle,water usually exists in the form of OH−anions and is stored in hydrous or nominally anhydrous minerals.[117,118]
The dominant mantle rock type,peridotite,can be hydrated in this way and the resulting phases can be understood by considering the ternary system of MgO–SiO2–H2O(dense hydrous magnesium silicates,DHMS).A secondary source of hydrous minerals can be found in the ternary system of Al2O3–SiO2–H2O(alumino silicate hydrates,ASH).The known phases in those two systems are shown in Fig.8.These phase diagrams feature a wide range of phases.Their water content is usually 50%or less,the exceptions being Al(OH)3and MgSi(OH)6(“3.65phase”).An overarching first-principles description of these phases is hampered by several problems.Several of these phases are likely to support water uptake across a range of compositions–see for example the sequence from“anhydrous phase B”via“phase B”to“superhydrous phase B”in the DHMS system,which is likely to have non-stoichiometric intermediates.Or consider nominally anhydrous minerals(such as olivine,Mg2SiO4)that can support water uptake through substitutional defects. Such scenarios are hard to capture using standard periodic boundary DFT calculations.On the other hand,almost none of the true ternary phases in either mineral system are known to undergo structural phase transitions under pressure;a curious situation quite unexpected as it is not in line with our knowledge about many other minerals or materials in general.Of course it is possible that all hydrous minerals decompose upon compression,leading to dehydration melting,before pressures are reached required to induce structural transformations.However,there is scope for CSP to test this hypothesis,because the complicated geochemistry involved in the formation of these minerals limits the experiments that can be performed on them;guidance from computational predictions would help in designing targeted experimental studies.
A successful example of computation aiding minerals discovery is the DHMS phase H,MgSiO2(OH)2, which was first constructed in calculations[119]and subsequently synthesised.[120]The crystal structure of phase H,with the very simple composition 1:1:1 of MgO,SiO2,and H2O,was guessed at by applying one of the ionic substitutions mentioned in the previous section,to a highpressure phase of aluminium oxide hydroxide,δ-AlOOH.The latter compound itself is a very stable mineral in the ASH system,in fact it is the hydrous mineral known to be stable to the highest pressure,at least 134 GPa in experiment.[121]A CALYPSO CSP study applied to AlOOH uncovered a new structural paradigm in a phase transition at pressure and temperature conditions applicable to super-Earth mantles.[122]As depicted in Fig.9,hitherto known AlOOH polymorphs feature corner-sharing regular AlO6polyhedra,with protons forming asymmetric and(at higher pressures)symmetric hydrogen bonds.The highest-pressure polymorph(the pyrite structure type)is expected to decompose into Al2O3and ice just below 300 GPa in the ground state. The structure found by CALYPSO substantially changes the structural arrangement,with irregular AlO7polyhedra and bent and asymmetric hydrogen bonds. This allows for much more compact packing and favours this phase over other AlOOH phases above 350 GPa. The trend towards relatively low-symmetry configurations with high coordination numbers is similar to that seen in ice in the multi-Mbar range.[62]The high-pressure P21/c phase of AlOOH is also relevant in other group 13 oxide hydroxides,and should appear at much lower pressures in GaOOH and InOOH;this study has stimulated further CSP explorations of the AlOOH phase diagram,with more highpressure phases being predicted.[123]

Fig.9.High-pressure crystal structures of AlOOH.(a)δ-AlOOH,(b)pyritetype AlOOH-Pa,(c)AlOOH-P21/c.Al(O,H)atoms are blue(red,pink)spheres,respectively.Al polyhedra and O–H–O hydrogen bonds are shown.
Another successful CSP study explored the high-pressure evolution of brucite,Mg(OH)2,which is arguably the simplest hydrous mineral in the DHMS system.[124]Brucite is the most important MgO–H2O binary phase and,apart from the“3.65 ˚A phase”the most water-rich phase in that mineral system. Its known crystal structure comprises layers of edge-sharing MgO6octahedra,with OH groups formed at every corner that arrange perpendicular to the layers. There are no hydrogen bonds in this structure,and their emergence under moderate pressures dominated the early high-pressure studies of this material.[125,126]Many metal hydroxides of the form M(OH)or M(OH)2form layered structures,[127,128]yet under pressure there are often phase transitions to threedimensional networks with rich hydrogen-bond topologies–CSP has been used successfully to identify these transitions and to corroborate(or even re-interpret)experimental structure solutions.[129–131]For Mg(OH)2,a similar transformation is predicted by CSP to take place(CALYPSO has confirmed these results):[124]the layered brucite structure becomes unstable under pressure and is superseded by a threedimensional network of corner-sharing polyhedra that is topologically equivalent to TiO2anatase(see insets in Fig.10),with OH groups forming very short hydrogen bonds in channels of the heavy atom network. This high-pressure polymorph delays the decomposition of Mg(OH)2into MgO and ice from 19 to 33 GPa in the ground state,making it a relevant water storage material inside the lower mantle.Figure 10 shows the P-T phase diagram of Mg(OH)2from DFT free energies based on the harmonic approximation but also including experimental estimates for a melting curve and for subduction slab geotherms.The figure shows that the high-pressure phase is close to stability in cold subduction slabs.As in other examples discussed above the CALYPSO results should be used to initiate AIMD simulations to quantify the high-temperature behaviour of these phases beyond the quasi-harmonic approximation. In this particular material,the transition from layered to three-dimensional polyhedral networks coincides with changing the constraints of protons from two-dimensional layers to one-dimensional channels;this could have significant implications for thermal and electrical conductivity based on diffusive proton sublattices.

Fig.10.P–T phase diagram for Mg(OH)2,which includes a parameterised melting curve(dashed black line),cold geotherms(red and blue lines)and ice melting curves(grey lines).Taken from Ref.[124].
The two examples discussed here present the simplest examples for a certain class of mantle minerals.CSP has been able to reveal intriguing phase transitions in both cases,and it stands to reason that its application to more complex mineral compounds will also yield new insights into high-pressure phases with new properties that are relevant in planetary interiors.
5.2.Helium reacting with ionic compounds
Mantle minerals are essentially ionic compounds,much of their stability driven by attractive and repulsive Coulomb interactions between ions of opposite or equal charges.A recent CSP study found that the smallest(and most inert)noble gas element,helium,readily forms compounds with ionic structures of unequal anion/cation numbers.[132]This“reactivity”of helium is not accompanied by the formation of any kind of chemical bonds but rather benefits from helium atoms lowering the Madelung energy of the ionic crystals when inserted between the majority ions. A one-dimensional schematic is shown in Fig.11: for simple AB compounds,insertion of helium will raise the Madelung energy,because favourable nearest-neighbour ionic interactions are lengthened. In contrast,for AB2compounds(or any ionic compounds AxBywith),there must be close contacts of the majority ions(this is especially true under pressure),but these are electrostatically costly and inserting helium between them will lower the Madelung energy. While the full 3D picture is clearly more complicated(and the p∆V term plays a role in any highpressure reaction)this simple idea led to the prediction of Hecontaining compounds AB2He with(A,B)in(Mg,F),(Ca,F),(O,Li),[132]and even(2e−,Na),the latter successfully interpreting the unusual Na2He compound[23]as an ionic compound of Na+cations and(2e)2−anions.If helium insertion can be stabilised according to the scheme in Fig.11 this would certainly apply to actual minerals as well.Indeed,helium has been found in experiments to penetrate amorphous and crystalline polymorphs of SiO2in significant quantities.[133–135]And,as discussed in Section 3,the iron oxide FeO2is predicted to take up helium in a FeO2He phase that is isostructural to Na2He.[22]
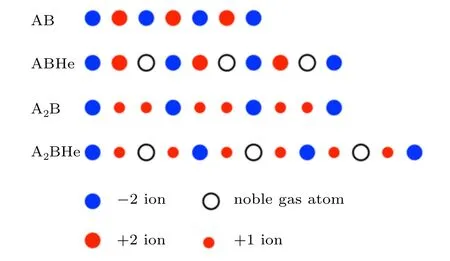
Fig.11. Schematic of He insertion into one-dimensional ionic compounds AB and AB2.Large blue(large red,small red)circles represent charges −2(+2,+1),white circles represent noble gas atoms.Taken with permission from Ref.[132].
6.Conclusion and perspectives
Crystal structure prediction has lots to offer to geo-and planetary sciences. This research field comes with unique challenges: chemical complexity,the role of high temperature,entropic stabilisation of solid solutions are but some of those.On the experimental side,direct measurements are difficult if not impossible,and laboratory setups need to be tightly constrained to be comparable to CSP calculations.However,across a variety of geological settings CSP has proven a very useful tool. It can be used to study compound formation of iron with volatile elements at planetary core conditions;can predict silicate-dominated minerals in rocky mantles;and help us understand the chemistry and physics inside icy planets.Some results from these areas,with the CALYPSO code heavily involved,were discussed in this review. There are more planetary scenarios where CSP could be applied:exoplanet research only begins to understand the types of planets that could form in other solar systems,and predicting the properties of their potential constituents(e.g.,carbon at multi-TPa pressures[136])is very useful to develop a better understanding of the formation and evolution of planetary systems and the place of our solar system amidst those.
There are recent methodological developments that suggest that CSP will become even more useful for geosciences in the future. A“geochemical coarsening”,based on automated learning of the relevant“building blocks”of minerals and other compounds under specific pressure conditions could lead to great acceleration of structure searching,which would afford an expansion of CSP into more complex chemical composition spaces. Recently,the CALYPSO package has been extended to train on-the-fly interatomic potentials using the Gaussian Approximation Potential(GAP[137])machine learning(ML)approach.[9]While not yet applied directly to the field of geosciences,the potential speedup of a combined DFT/ML approach holds promise for the future.An interesting question remains on how CSP can be used at finite temperatures,i.e.to produce realistic free energies of materials.It is possible that ML can help with this as well,e.g.by constructing cheap interatomic potentials that allow on-the-fly calculations of dynamical properties and thus vibrational entropies.
Acknowledgment
The author is indebted to collaborators both from the crystal structure prediction and the geoscience community for continuous discussions,including Prof.Y.Ma and Dr.Y.Wang(Jilin University),and Prof.M.Mookherjee(Florida State University).A Research Fellowship for International Young Scientists by the National Natural Science Foundation(NNSF)on“In-silico studies of planetary materials”is gratefully acknowledged. Computing resources provided by the UK national high performance computing service,ARCHER,and the UK Materials and Molecular Modelling Hub,which is partially funded by EPSRC(EP/P020194),and for which access was obtained via the UKCP consortium funded by EPSRC grant No.EP/P022561/1,are gratefully acknowledged.

- Chinese Physics B的其它文章
- Compact finite difference schemes for the backward fractional Feynman–Kac equation with fractional substantial derivative*
- Exact solutions of a(2+1)-dimensional extended shallow water wave equation∗
- Lump-type solutions of a generalized Kadomtsev–Petviashvili equation in(3+1)-dimensions∗
- Time evolution of angular momentum coherent state derived by virtue of entangled state representation and a new binomial theorem∗
- Boundary states for entanglement robustness under dephasing and bit flip channels*
- Manipulating transition of a two-component Bose–Einstein condensate with a weak δ-shaped laser∗