
Recent progress on the prediction of two-dimensional materials using CALYPSO
2019-11-06 00:46ChengTang唐程GurpreetKourandAijunDu杜爱军
Chinese Physics B 2019年10期
Cheng Tang(唐程),Gurpreet Kour,and Aijun Du(杜爱军)
School of Chemistry,Physics and Mechanical Engineering,Queensland University of Technology,Gardens Point Campus,QLD 4001,Brisbane,Australia
Keywords:CALYPSO methodology,two-dimensional materials,structural prediction
1.Introduction
Throughout history,the exploration of unknown territory greatly attracts the interest of researchers,however it is really a time intensive process in novel materials discovery.In order to shorten such processes,computational methods have been regarded as one of the most important approaches in materials science.[1]Several structure prediction methods are developed based on the exploration of potential energy surface(PSE),[2]including data mining,[3]minima hopping,[4]genetic algorithm and energy minimisation,[5,6]basin-hopping,[7]and particle-swarm optimization(PSO).[8,9]Therein,Crystal structure AnaLYsis by PSO(CALYPSO)is a highly efficient package in materials design and prediction,especially under high pressure.[10,11]Up to now,a large number of functional bulk materials have been discovered by CALYPSO method,involving lithium batteries with high energy density,[12–14]superconductors,[15–17]photovoltaics,[18,19]electronics,[20,21]superhard[22–24]and deep-earth materials.[25,26]Additionally,CALYPSO methodology also plays an important role for structure prediction in low dimensional materials.
The discovery of graphene opens the gate for twodimensional(2D)materials,[27]which typically have a thickness of a few atomic layers.[28]For example, graphene shows quantum Hall effect,high carrier mobility,and electronic chirality,owing to its unique 2D honeycomb crystals and massless Dirac dispersion.[29–31]In addition to graphene,a variety of 2D sheets with novel structures have been discovered theoretically and experimentally,including transition metal dichalcogenides(TMDs),[32,33]black phosphorus(BP),[32,34,35]MXene,[36]metal–organic frameworks(MOFs),[37]and heterostructures.[38–40]These 2D materials exhibit extraordinary electronic,optical,and magnetic properties,leading to their great potentials in electronics,optoelectronics,spintronics,valleytronics,magnetronics,catalysis,etc.Besides,experimental discovery of new 2D materials is a real difficulty.Although several methods,such as mechanical exfoliation[41,42]and chemical vapour deposition(CVD),[43]are well-developed,it is still difficult to fabricate high-quality atomic layers.Besides,due to the large number of possible 2D structures,such trial-and-error experiment without any guidelines and instructions is a waste of time and efforts.Therefore,theoretical prediction and design would significantly decrease the expense and accelerate the discovery of new 2D materials with novel properties.
To date,CALYPSO,as one of popular methods for structure prediction,has already achieved great success in the field of designing novel 2D materials. Based on statistical data,figure 1 displays the evolution of publications on 2D structure prediction by CALYPSO since 2011. It shows a rapid rise in the number of publications over the years and it is expected to increase further in the future.However,the lack of an overview on the recent advances,perspectives,and challenges in the field of structure prediction on 2D materials greatly restricts the discovery of new materials both in experimentally as well as theoretically.Thus,in this review,we retrospect the recent achievements of CALYPSO methodology in structural design and prediction of novel 2D materials.Furthermore,we also conclude the challenges and perspectives on the basis of recent progress for further development of 2D structure predictions by CALYPSO methodology.
2.CALYPSO methodology
As a stochastic global optimization method,PSO was first proposed in 1990 s by Kennedy and Eberhart.[9]Then,through combining the advantages of PSO method with other important techniques,CALYPSO methodology has been developed by Wang and Ma[9]et al.Several publications have already elucidated the theory of CALYPSO methodology in detail,[8,9,44–46]herein,we give a brief summary on such method of predicting the structure of 2D materials.Generally,the PSO algorithm in CALYPSO software for crystal prediction contains four main steps:i)generation of random structures,ii)local optimization,iii)identification of unique local minima,iv)generation of new structures. In order to make it efficient and reduce computational costs on structural design and prediction,CALYPSO also integrates several critical techniques,including structural evolution,structural characterization techniques,symmetry constrains,and local structural optimization,as amply described in pioneering works.[8]Recently,CALYPSO methodology has been widely used to predict 3D,2D,and 0D-isolated materials and achieved great success in this field.[9]
Among them,a 2D structure search module was developed in CALYPSO method for efficiently and accurately deal with few-layer structure predictions.[47]The general flow chart for 2D materials prediction is illustrated in Fig.2(a)with above mentioned four main steps.[9]In such module,the generated initial structures are constrained by the 17 in-plane space groups,which will filter out various inappropriate random structures.As shown in Fig.2(b),a vacuum level is automatically generated in 2D structures to avoid the interactions between neighboring layers. Besides,the in-plane inflection can be tuned by a distortion parameter(∆z)not only in monolayer but also for a multi-layer system,and the van der Waals interaction is introduced to multiple layers. It is worth mentioning that layered heterostructures can also be generated by controlling the atomic categories and ratios of each layer(Fig.2(c)). Then,in order to get the local and global minimum of basin,all of the generated structures are examined by other ab initio or force-field based total energy packages,which have been interfaced with CALYPSO code,including VASP,[48]SIESTA,[49]Quantum-Espresso,[50]and CASTEP.[51]
3.Novel 2D materials discovery via CALYPSO
Since 2D structure search module in CALYPSO code was first introduced by Wang and Ma et al.in the early 2010 s,[47,52–54]a variety of novel 2D structures at given chemical compositions are designed on the basis of this methodology and various extraordinary properties are discovered aswell,leading to their wide applications in electronics,optoelectronics,magnetronics,spintronics,and photovoltaics,as summarised in Table 1.In this section,we comprehensively review the current achievements of 2D structure prediction on both elements and compounds.

Table 1.Novel Properties of 2D structures predicted via CALYPSO method.
3.1.Elements
Graphene is the first discovered 2D materials with perfect honeycomb structure. Such configuration of hexatomic rings provide strong in-plane coupling of π electrons(sp2hybridization), which is beneficial to the stability of 2D structures. In addition to graphene,several 2D carbon allotropes which consists of quadrilateral,pentagonal,hexagonal,octagonal,and decagonal atomic rings are designed through CALYPSO.By adding the non-hexagonal rings in graphene,carbon allotropes exhibit different electronic properties,including metallic,semi-metallic,semiconducting as well as insulating. In 2014,Ma and Miao et al.discovered number of carbon allotropes with low energies by using structure searching methods(Fig.3(a)),which can also be derived from graphene through different artificial geometry indexes(Ising-like model),as shown in Fig.3(b).[57]Among them,S-graphene,D-graphene,and E-graphene show Dirac points at Fermi levels,indicating the common feature of Dirac dispersion beyond graphene. Besides,pza-C10[55]and H-net[99]carbon nanosheets are semiconducting,whereas,ψ-graphene,[100]DHP-graphene,[101]and net-τ carbon allotropes[102]are metallic. It is worth mentioning that these metallic carbon allotropes also show potential application in Lithium-ion batteries with low diffusion energy barrier and open circuit potential.

Fig.3. (a)Relative energies of new carbon structures obtained by CALYPSO.[57](b)Ising-like model for derived carbon layers from graphene.[57](c)Predicted structures of stable 2D borophene. Red and yellow balls donate boron atoms moving outward or inward from the plane,resulting in buckled boron sheets.[53](d)A and B site in 12×12 graphene supercell and the pair-interaction energy between nitrogen atoms as a function of the pair distance from the DFT total-energy calculations for the relaxed structures.Numbers are the carbon-lattice sites for nitrogen substitution.[108]Reprinted with permission from Ref.[57],Copyright 2014 Royal Society of Chemistry.Ref.[53],Copyright 2012 American Chemical Society.Ref.[108],Copyright 2012 American Physical Society.
Boron with one electron less than carbon is the second common element in forming low-dimensional materials.Over 9000 boron sheets have been predicted by CALYPSO at ambient condition,and part of them are proved to be dynamically stable by phonon calculations(see Fig.3(c)).[53,103]Different from graphene,2D borophene favours buckled triangular lattice with discontinuous arrangements of hexagonal holes,due to the electron deficiency. Among them,most of boron sheets show metallic properties,intriguingly,double Dirac cones with Fermi velocity higher than graphene are predicted in P6/mmm boron sheet.[60]These predicted boron sheets are expected to advance further investigations and understandings on 2D boron-based compounds. Importantly,some of the predicted 2D borophenes have been synthesized in following experiments.[104,105]Furthermore,several 2D allotropes of selenium,[106]germanium,[107]and silicon[66]are also discovered.
In addition,CALYPSO package can also be readily interfaced with other algorithm or technology to meet the requirements of different conditions. In order to study the effect of micro-porosity on graphene-like materials,Miao et al.developed the PSO and the gene mutation mixed algorithm to randomly generate holes in initial structures.[109]The formation energies of C allotropes increase quickly with the micro-porosity in a parabolic relationship,while the electronic properties also depend strongly on the porosity in their calculations. In contrast,boron allotropes show a weaker and linear relationship on micro-porosity. Additionally,figure 3(d)shows two semiconducting N-doped graphene structures with high stability,C3N and C12N,which are generated through combining the PSO and cluster-expansion technique in 2012.[108]Their work reveals the strong electrostatic repulsion between nearest-neighboring nitrogen atoms,which prevents the full nitrogen-carbon phase separation in N-doped structures.
3.2.Metal-free compounds
In recent years,2D metal-free nanosheets has withdrawn considerable attentions both in experiment and theory.After continuous efforts,several of them,such as C3N4and BN,can be fabricated in large scale as well as high quality,leading to promising potentials for wide applications.[110–112]Via CALYPSO methodology,more than thirty metal-free nanosheets are predicted to be stable.Among them,most widely studied nanosheets are carbon-and boron-based binary compounds,such as SiC,[56]PC6,[83,113]SiB[114]and B2S2.[82]Additionally,metal-free binary compounds based on other non-metal elements(such as Si,N,P,and O)have also been discovered,forming some special 2D structures(SixOy,[85,115]SixPy,[72,77]SiN,[116]and PN[117]sheets).Besides,a few ternary(B6C2P2and BxCNy),[118–121]and quaternary(SiBCN)[122]2D structures are also predicted,leading to their applications in electronic and optoelectronic devices.

Fig.4.Predicted low-energy 2D structures and relative formation energies for(a)BxCy and(b)SixC1−x.[52,62]Reprinted with permission from Ref.[52],Copyright 2011 American Chemical Society.Ref.[62],Copyright 2017 Royal Society of Chemistry.
3.2.1.Carbon-based compounds
The versatile configurations of hybridization(sp,sp2,and sp3) for carbon make it the most feasible atom to bond with other non-metal atoms. Most predicted boron carbides are metallic, whereas, B2C and BC3are semiconducting.[52,123,124]In 2011,Xiang et al.discovered several unexplored 2D boron-carbon compounds under different stoichiometry,including BC5,BC3,BC2,BC,B2C,B3C,B5C(as shown in Fig.4).[52]For C-rich conditions,boron forms 1D zigzag chain(except for BC3)in the most stable structures as B-doped graphene,while for B-rich compounds,a novel planar-tetracoordinate carbon motif with an approximate C2vsymmetry is observed. Since silicon has the same number of valence electrons as that of carbon,the former expected to form stable monolayers in a large range of stoichiometric compositions with carbon.[56,62,125,126]Figure 4(b)shows the formation energies of predicted SixC1−xunder conditions from C-rich to Si-rich.Among them,three stable structures(t-SiC,t-Si2C,and γ-silagraphyne)indicate the diversity of bonds(Si–C,Si–Si,and C–C)in SixC1−x.[62]Furthermore,C3N4has been experimentally fabricated and widely studied so far,which inspires the exploration of other 2D structures containing carbon and nitrogen(phosphorus)elements.Consequently,predicted structures of P3C and PC6show buckled hexatomic rings,[83,127]while C2P4,C4N,and C4N4mix with multiple atomic rings.[64,128,129]Moreover,two unique 2D carbon dioxides are also discovered,exhibiting a negative Poisson’s ratio and a huge band gap.[88]
3.2.2.Boron-based compounds
Boron, as electron-deficient non-metal atoms, can be strongly bonded with other non-metal atoms in lowdimensional limit. Except for above mentioned BxCy,other binary compounds, such as BH,BxSiy, BxPy, BxSy, and porous BO,[58,69,74,82,86,114,130–136]are also predicted by CALYPSO method during these years. Boron can form novel structures with silicon under different stoichiometric compositions,through the planar sp2hybridization.[114]However,as shown in Fig.5(a),the buckled hexatomic rings exist in low-energy structures of BP3,whereas,it becomes pentagonal rings as the number of phosphorus atom increases from three to five.[74,134]The electron localization function(ELF)shows that all the B and P atoms undergoes sp3hybridization,indicating their semiconducting properties.Additionally,three boron sulfied compounds are predicted through CALYPSO method.However,the electronic properties of B3S(metal),B2S(Dirac semimetal)and B2S2(wide-bandgap semiconductor)are completely different,which mainly ascribe to the different bonding characters.For B-rich BS compounds,B and S atoms are covalently bonded together,especially,strong sp2hybridization exists in B2S,leading to its Dirac dispersion.[69,135]However,in the case of B2S2(Fig.5(b)),it shows ionic characters between B2pair and S atoms,suggesting their significant electron transfer.Therefore,the strong interaction between B2pair and S atom makes it an intrinsic semiconductor,which is comparable to 2D MoS2.[82]The ionic character of boron is considerable for designing new 2D metal-free nanosheets for future electronics.

Fig.5.(a)Predicted structures of BP3 and BP5.[74,134]The ELF of penta-BP5 monolayer are displayed with the isovalue of 0.85.(b)The structures of B2S2 and band charge density for highest valence and lowest conduction.The ionic B2 pairare shown as reference.[82]Reprinted with permission from Ref.[74],Copyright 2017 Royal Society of Chemistry.Ref.[134],Copyright 2017 Nature Publishing Group.Ref.[82],Copyright 2019 Royal Society of Chemistry.
3.3.Metal-containing compounds
In addition to above mentioned 2D materials,metalcontaining structures have also been widely studied,such as transition metal dichalcogenides(TMDCs)and MXenes,which exhibit extraordinary electronic and optical properties and play an important role in a variety of applications,including field-effect transistors(FETs),energy storage,catalysis,and solar cells.In general,metal-containing nanosheets usually possess robust charge transfer from metal to non-metal atoms,due to their significantly different electronegativity.Hundreds of stable metal-containing compounds have been predicted through CALYPSO methodology,including binary(hydrides,borides,carbides,silicides,nitrides,phosphides,oxides,and sulfides)and several ternary compounds.
3.3.1.Binary compounds
Hydrides. Hydrogen(H)is the only non-metallic element which has just one electron in its outermost shell,thus,H favours to be an electron donor rather than an electron acceptor.To the best of our knowledge,there is still no report on experimentally fabricated 2D metal hydrides. However,with the development of structural prediction,many metal dihydride monolayers are discovered,including BeH2,ScH2,TiH2,VH2,CrH2,FeH2,CoH2,NiH2.[87,95]Such a new family of MH2monolayers are promising functional materials for electronics and spintronics.It is also expected to predict and fabricate more metal hydrides under different stoichiometry in the future.
Borides. Metals can form bonds with boron atoms under different stoichiometric compositions in 2D limit,including MB,MB2,MB3,MB4,and MB6,due to the variable orbital hybridized configurations of boron(as shown in Fig.6).[61,70,71,98,137–143]In 2016,Zhang et al.first discovered three planar FeB6phases with hypercoordinate Fe atoms locating at the center of six-or eight-membered boron rings,forming highly stable graphene-like structures.Here,strong electron transfer from Fe to B is found to be a main reason for their dynamical stabilities.[142]Most recently,other two stable MB6(M=Co,Ni)structures are found by examining all of the possible structures of MxBy(x,y=1–6)monolayer.[70]It is worth mentioning that 2D MB6have two feasible synthesis routes,including depositing metal atoms on the δ4boron sheet and direct chemical growth. Furthermore,Dirac state FeB2monolayer and planar TiB4are also predicted by decreasing the stoichiometric composition of B atoms.[61,139]Since discovered 2D metal borides are mainly concentrated in transition metals,following works could pay more attention to the main group metals,which may find out new properties and applications.

Fig.6.(a)Electron localization function(ELF)of FeB6 with the isovalue of 0.3 a.u.and its 2D maps sliced perpendicular to(001)direction.[142](b)Optimized structures for NiB6 and CoB6 and the ELF map for NiB6.[70,71](c)Predicted structure for planar FeB2.[61](d)ELF and structure of TiB4.[139]Reprinted with permission from Ref.[142],Copyright 2016 American Chemical Society.Ref.[71],Copyright 2019 Royal Society of Chemistry.Ref.[61],Copyright 2016 American Chemical Society.Ref.[139],Copyright 2017 Royal Society of Chemistry.
Carbides. A large number of metal carbides have been predicted theoretically, exhibiting extraordinary electronic,magnetic,and optical properties.[54,63,65,78,84,89,90,144–152]In 2014,Be2C with quasi-planar hexacoordinate carbons was first predicted by CALYPSO code,which opens the gate for designing 2D MxCy(M=metal element)monolayers.[144]In these predicted metal carbides, metal atoms are ionically bonded with carbon atoms,due to the significant electron transfer between them. Interestingly,a novel 2D TiC3monolayer possesses alternant zigzag Ti chain and n-biphenyl unit as shown in Fig.7(a). In such structure,the nearestneighboring C atoms are covalently bonded,while adjacent Ti–Ti and Ti–C bonds are metallic and ionic,respectively.[152]This unique structure not only shows remarkably high storage capacity of Na atoms but also low barrier energy and open-circuit voltage,rendering it a promising anode material for sodium-ion batteries. Also,silicon being the group member of carbon,possesses great potentials in forming novel planar structures with metals.[67,153,154]On the basis of CALYPSO,Wang et al.discovered three stable 2D structures of TixSiy(Ti2Si,TiSi2,TiSi4as shown in Fig.7(b)),suggesting applications in magnetonics,catalysis,and superconductor,respectively.[97]
Nitrides.Nitrogen is feasible to form ionic bonds with metal atoms,due to its strong electronegativity and unpaired p electrons.Recently-predicted 2D metal nitrides exhibit variety of planar structures,such as quadrilateral CrN,[155]penta-PdN2,and PtN2,[156]graphene-like Be3N2,[157]Be2N6,[73]and Mg3N2,[158]and buckled YN2[159]structures. Most of them are semiconductors with moderate bandgaps,and CrN shows robust ferromagnetism. In addition to nitrides,other non-metal elements in this group can also form stable 2D ionic structures,including half-metallic MnP and MnAs,[96]semiconducting SnAs,[160]and GexPy[76]and Dirac semimetal BeP2[161]monolayers.Figure 7(c)displays the convex hull of the formation enthalpy for all predicted structures of GexPy(GexAsy),containing the global and local minimum of each concentration.These monolayers possess variety of geometric structures under different concentrations and exhibit completely different electronic and optical properties.[76]Notably,the elements in group 15 change from non-metal to metal with the increase of the atomic radii,due to the weak elastic Coulomb interaction between nucleus and outermost electrons.Therefore,it is highly possible to form ionic 2D structures between these elements of the same group. Till now,several 2D ionic compounds are predicted with stable BP-like structures,such as BiSb,SbN,and BiP.[93,162]

Fig.7.(a)Zigzag Ti chain and n-biphenyl structural unit in TiC3 monolayer and its ELF maps.[152](b)Cohesive energies of predicted Ti1−xSix monolayers and three stable structures under different composition.[97](c)Formation enthalpy versus concentration(y)for GexPy and stable structures.[76]Reprinted with permission from Ref.[152],Copyright 2018 American Chemical Society.Ref.[97],Copyright 2016 American Chemical Society.Ref.[76],Copyright 2018 American Chemical Society.
Sulfides.To date,1 T-,2 H-,and 1T’-MoS2have attracted much attention,due to their wide applications in electronics and photocatalysis. Based on CALYPSO methodology,two newly metastable MoS2monolayer are discovered(Fig.8),namely 1 Td-andMoS2.[59,163]Their electronic properties are totally different,1 Td-MoS2is a semiconductor monolayer with a bandgap of above 1 eV,whereas,MoS2shows topological Dirac dispersion under 2%external strains.In addition to MoS2monolayer,many stable 2D metal sulfides are predicted within different structures,including H-MoS2-like Au6S2,[75]MXene-like Li2S,[164]rugged pentagonal PdS2[165]and other special Co2S2and CuS monolayers.[81,166]Moreover,as oxygen,belong to the same group as that of sulfur, it can also form a variety of structures with metal elements.[167–169]In 2019,Zeng et al.introduced 18 functional monolayer metal oxides with wide bandgaps,superior oxidation resistance and ultrahigh carrier mobilities.[80]
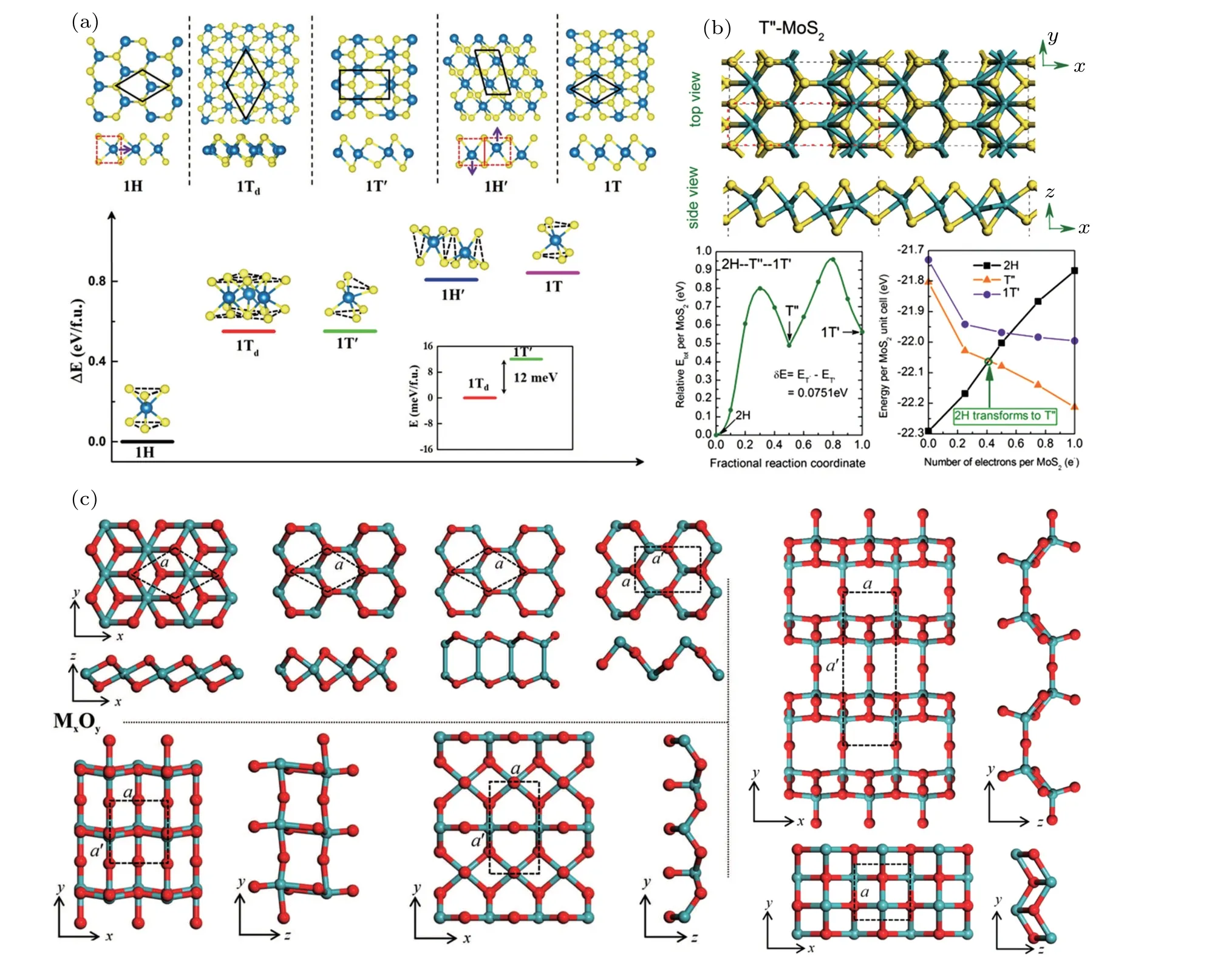
Fig.8.Structures and relative energies of(a)1 Td-MoS2 and(b)MoS2,respectively.[59,163](c)Atomic structures of monolayer metal oxides,including ZrO2,HfO2,NiO2,PtO2,GeO2,SnO2,MoO2,WO2,SnO2,MoO3,TiO2,Mo2O5 and W2O5.[80]Reprinted with permission from Ref.[59],Copyright 2016 Royal Society of Chemistry.Ref.[163],Copyright 2018 Royal Society of Chemistry.Ref.[80],Copyright 2019 Royal Society of Chemistry.

Fig.9.(a)Predicted structures of AuGaS2 and bandgaps of extended AuMX2 monolayers.[79](b)Different alloyed configurations of CrWI6 and CrWGe2Te6,and their relative energies.[92](c)The structures of most stable VSSe and their phonon spectra.[94]Reprinted with permission from Ref.[79],Copyright 2018 American Chemical Society.Ref.[92],Copyright 2018 American Chemical Society.Ref.[94],Copyright 2019 American Chemical Society.
3.4.Ternary compounds
As we all know,2D structures containing more than two elements are usually hard to be predicted in theory,due to the complicated interactions and variably stoichiometric compositions among them.Since 2017,several 2D ternary compounds are theoretically discovered on the basis of CALYPSO,such as metallic Ti3BN,[170]semiconducting AuMX2(M=Al,Ga,In,X=S,Se),[79]and ferromagnetic CrWI6,CrWGe2Te6,[92]and VSSe.[94]In Fig.9(a),AuGaS2exhibits three stable phases(α,β,and γ),which can be extended to other AuMX2monolayer through the elemental replacement within the same group in the periodic table.[79]Herein,all of the AuMX2are semiconductors with a bandgap of over 2.0 eV.Besides,CrWI6and CrWGe2Te6are derived from the recently fabricated 2D monolayer CrI3and CrGeTe3by forming different alloyed configurations in a 2×2 supercell(Fig.9(b)).[92]Here,structure prediction based on CALYPSO confirmed the honeycheckboard(HC)pattern is the most stable one among all the possible alloyed configurations. Moreover,CALYPSO also can be used to predict the Janus structures such as VSSe as shown in Fig.9(c),1T-and 2H-VSSe are thermally and dynamically stable and exhibit excellent multiferromagnetic properties.[94]Although CALYPSO has made some progress in the discovery of ternary compounds,the 2D structure prediction on multi-elements is a persistent challenge.
4.Summary and perspective
As summarized in this review,a large number of novel 2D structures with intriguing properties have been predicted through recently-developed CALYPSO methodology and firstprinciples simulations.After a brief introduction of the theoretical background,we then comprehensively retrospect the predicted 2D materials,including elements,metal-free and metal-containing compounds,exhibiting great potentials in electronics,optoelectronics,magnetronics,spintronics,and photovoltaics.Therefore,CALYPSO methodology has made great achievements in novel 2D materials design.The discovered 2D structures and their extraordinary properties are also expected to instruct and interpret further experiments.
CALYPSO package shows many advantages in 2D structure prediction. First,it is highly efficient through combining PSO algorithm and several enhanced techniques.Second,based on both global and local PSO algorithms,CALYPSO can deal with more complex systems and guarantee its validity. Besides,in order to avoid the possibly premature convergence at local minimum,a certain number of new structures are randomly generated,ensuring the structural diversity.Third,CALYPSO is a user friendly and convenient package.For 2D structural prediction,the chemical composition,layer numbers and distance of layer gaps are necessary to be set for a given compound.Thus,CALYPSO package can be widely used to design multifunctional materials under different conditions.Fourth,not only the most stable structure but variety of metastable structures are discovered under each stoichiometric composition.Some of these“metastable”structures also possess highly dynamical and thermal stability,which could lead to phase transitions at a certain conditions. Fifth,it is convenient to interface CALYPSO with other algorithm or technologies to meet the requirement for designing some special structures(such as microporosity and dopant).Therefore,CALYPSO is an irreplaceable tool for 2D structure prediction.
Despite the great success on 2D structure prediction,several challenges still exist in this field,requiring further development of CALYPSO.First of all,the extreme conditions,such as high pressure and temperature,are a persistent problem in experiments,due to the restriction of devices and huge cost for trail-and-error tests. Thus,theoretically structural design is eagerly expected to guide the experiments in such extreme conditions. Although CALYPSO has made some progress on 3D structure prediction under extremely high pressure,it is still hard to be extended to 2D structures,mainly relying on the existence of vacuum layers.Compared to 3D crystals,2D limited sheets should be separated by the vacuum layer to avoid the interactions by neighboring atomic layers. However,this vacuum layer allows such 2D atomic layers to shield the pressure along this direction as well,that would further influence the effect of the given pressure.Therefore,it is impossible to obtain the accurate 2D structures under high pressure through current techniques.Besides,temperature also plays an important role for materials designing.However,recently structural searching methodologies are all restricted to obtain the free energy landscape at 0 K,leading to a huge gap between theoretical prediction and experimental synthesis.In addition,the phase transition between the stable and metastable structures could happen under different temperatures,therefore,it is worthy to explore whether or not one certain phase could be synthesized at a given temperature in experiments.Unfortunately,no methods or techniques can address these problems so far.
Another challenge is 2D structural design for large scale systems and multilayer systems. Although CALYPSO has successfully predicted several 2D structures with ternary and quaternary elements,it is impossible to predict other 2D structures with a large number of atoms in one unit cell,such as metal-organic frameworks,covalent organic frameworks and multi-element alloy.Such large systems are hard to be dealt with by recently structure prediction methods. In such system,not only the significantly growing computational time is a huge challenge,but also the structural diversity cannot be guaranteed.Therefore,new efficient searching algorithm for large scale systems is urgent to be developed based on symmetry constraints and current structural databases.
Acknowledgment
We acknowledge the financial support by Australian Research Council under Discovery Project(Grant No.DP170103598)and computer resources provided by highperformance computer time from computing facility at the Queensland University of Technology,NCI National Facility,and the Pawsey Supercomputing Centre through the National Computational Merit Allocation Scheme supported by the Australian Government and the Government of Western Australia.

- Chinese Physics B的其它文章
- Compact finite difference schemes for the backward fractional Feynman–Kac equation with fractional substantial derivative*
- Exact solutions of a(2+1)-dimensional extended shallow water wave equation∗
- Lump-type solutions of a generalized Kadomtsev–Petviashvili equation in(3+1)-dimensions∗
- Time evolution of angular momentum coherent state derived by virtue of entangled state representation and a new binomial theorem∗
- Boundary states for entanglement robustness under dephasing and bit flip channels*
- Manipulating transition of a two-component Bose–Einstein condensate with a weak δ-shaped laser∗