
2-脱氧链霉胺取代衍生物的结构确证和1H核磁定量
2019-12-03 09:24王海东苏晓春代博娜倪瑶李红杰钱秀萍
中国抗生素杂志 2019年11期
王海东 苏晓春 代博娜 倪瑶 李红杰 钱秀萍,*
(1 常州方圆制药有限公司,常州 213125;2 上海交通大学分析测试中心,上海 200240;3 上海交通大学药学院,上海 200240)
抗菌药物一直被作为临床感染性疾病治疗的重要手段,但由于抗菌药物合理使用问题的日益突出,耐药菌株引起的感染是感染性疾病高病死率的主要原因[1]。新型氨基糖苷类抗生素如巴龙霉素(paromomycin)、西索米星(sisomicin)、依替米星(etimicin)、奈替米星(netilmicin)、阿米卡星(amikacin)、阿贝卡星(arbekacin)、妥布霉素(tobramycin)等对耐药菌感染具有积极的治疗作用[2-4]。在阿米卡星合成和工艺改进中有两个重要的中间体化合物1和化合物2需要进行结构确证和定量分析,然而化合物1和化合物2无紫外吸收,且无商品化的标准品。氨基糖苷类抗生素的含量检测有HPLC、荧光法、离子色谱法、流动注射化学发光法等[5]。常用的高效液相色谱法(HPLC)需要对样品进行衍生化处理或采用联用技术等复杂操作,商品化标准品的缺乏更给定量分析带来了困难[6]。氢核磁振定量分析(quantitative nuclear magnetic resonance,qNMR)集物质鉴别和含量测定于一体已应用于新药研发、对照品研究和质量控制等药学领域[7-9],使用单一参照化合物即可定量多个组分,而且比值定量测定不需要强度校正,精确度和精密度好,因此对药物研发过程中的混合物及含杂质样品的定量分析具有独特的优势[9]。本论文采用13C NMR、1H NMR、1H1H-COSY、HMBC和MS对化合物1和化合物2进行了结构确证,并应用1H NMR内标法测定了其量,为阿贝米卡星的工艺改进提供依据。
1 材料与方法
1.1 仪器与试药
Bruker AvanceⅢ 600MHz超导核磁共振波谱仪(瑞士,Bruker Biospin公司);岛津-LC-20AT HPLC色谱仪(日本,岛津);Mettler Toledo XP6电子天平(瑞士,Mettler Toledo公司);赛默飞ThermoScientific LTQ orbitrap XL高分辨液质联用仪。
重水(D2O,规格0.5mL,99.9% atom% D)购自Cambridge Isocope Laboratories;马来酸(TraceSure®)购自WAKO和光纯药工业株式会社;甲酸钠(纯度99.998%)、冰乙酸(HPLC级)、无水硫酸钠(HPLC级)购自Aladdin;硫代乙醇酸(HPLC级)、邻苯二甲醛(HPLC级)、戊烷磺酸钠(HPLC级)购自TCI;乙醇(HPLC级)购自TEDIA;甲醇(HPLC级)购自Merck;氢氧化钠(药检专用)购自沃凯;硼酸(GR级)购自国药集团化学试剂;水为高纯水。化合物1和化合物2由常州方圆制药有限公司合成。
1.2 1H NMR定量测定
供试溶液配制:准确称量化合物1和化合物2约4~22mg、内标物马来酸和甲酸钠约6.5mg于2mL EP管中,加入D2O 0.6mL,超声(35kHz)10min使其完全溶解,再转移适量至5mm的核磁管中备用。
仪器参数设置:采用zg30脉冲序列在恒温(25℃)下获取1H NMR谱。具体实验参数:谱宽(SWH)12019.23Hz,采样点数(TD)64K,采样时间(AQ)2.73s,驰豫延迟时间(D1)20s,采样次数(NS)16次,空扫次数(DS)2次,探头温度298K。在上述NMR采集条件下采集信号,进行相位调整和基线校正后,测定化合物1和化合物2以及对应内标物定量峰面积的相对比值,采用公式(1)计算化合物含量。
式中:P为待测样品纯度(%);As为样品定量峰的积分面积;ns为样品定量峰代表氢原子数;Ms为样品相对分子质量;ms为样品质量(g);Ar为内标峰的积分面积;nr为内标峰所代表氢原子数;Mr代表内标的相对分子质量;mr内标质量(g);Wr为内标物的百分含量。
1.3 邻苯二甲醛柱后衍生化HPLC分析
色谱柱:ZORBAXElipse XD B-C18( 250mm×4.6mm,5μm);流动相A:0.025mol/L戊烷磺酸钠溶液加0.1mol/L无水硫酸钠水溶液(乙酸调pH3.5),流动相B:甲醇;洗脱程序为流动相A:流动相B:95:5(V/V);流速:0.9mL/min;检测波长:330nm;柱温:40℃;进样体积:10μL;衍生化试剂:称取15.6g氢氧化钠和24.72g硼酸水溶解后加入4%邻苯二甲醛乙醇溶液,水稀释至2L,再加入2.0mL硫代乙醇酸混匀;衍生化试剂流速:0.6mL/min;衍生化温度:55℃。
1.4 LC-LC/MS分析
色谱柱: Agilent ZORBAXSB -C18(4.6mm×250mm,5μm);流动相A:0.3mol/L三氟乙酸水溶液,流动相B:乙腈;洗脱程序为流动相A:流动相B:96:4(V/V);进样体积20μL;柱温40℃;流速:0.9mL/min;N2流速:2.0L/min;正离子扫描30min;扫描范围:50~2000(m/z)。
2 结果
2.1 化合物1和化合物2的邻苯二甲醛柱后衍生化HPLC分析
对化合物1和化合物2采用邻苯二甲醛柱后衍生化HPLC分析,结果见图1。化合物1除了保留时间Rt14.47min的主峰P1外,还含有至少5个杂质,杂质峰a、b、c、d、e的HPLC保留时间分别为Rt9.05、12.05、12.79、16.39和17.55min。化合物2除了Rt 12.17min的主峰P2外,也至少含有a'、b'、c'、d'、e' 5个杂质峰,Rt分别是7.71、8.91、10.67、13.2和15.32min。化合物1的色谱纯度(n=3)为95.97%±0.16%,化合物2的色谱纯度(n=3)为97.58%±0.02%,结果见表1。
2.2 NMR分析的溶剂选择以及化合物1和化合物2的核磁谱图归属和质谱分析
本实验针对NMR常用溶剂进行实验,发现化合物1、化合物2以及内标物在D2O有较好的溶解性,而且溶剂峰与样品、内标物的吸收峰没有发生重叠,因此选择D2O做溶剂。
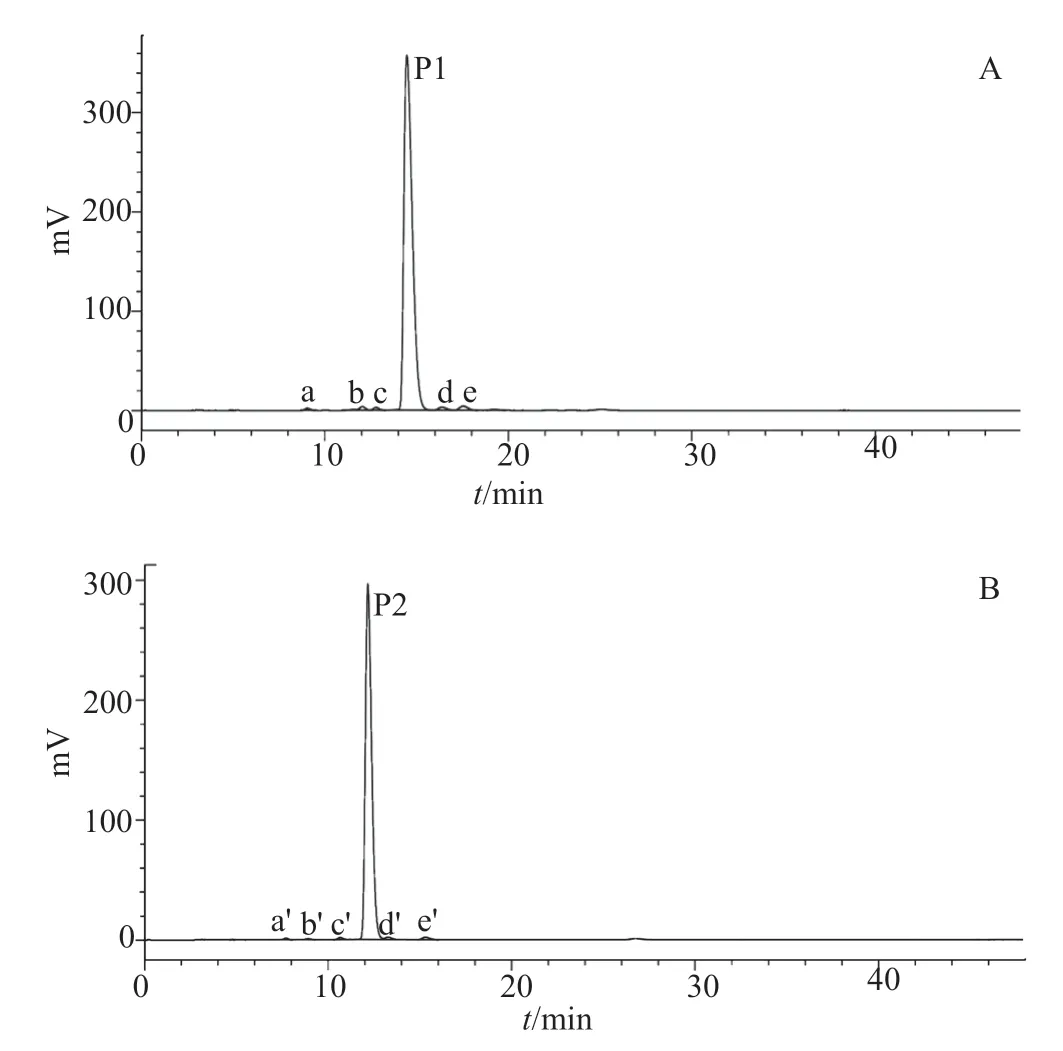
图1 化合物1和2邻苯二甲醛柱后衍生化HPLC色谱图Fig.1 HPLC chromatograms of compounds 1 and 2 with postcolumn derivatization of o-phthaldialdehyde
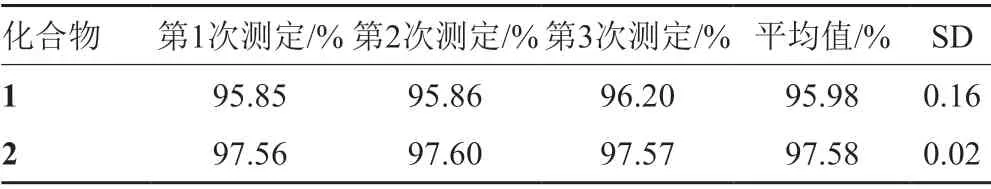
表1 化合物1和2的HPLC纯度测定Tab.1 Determination of compounds 1 and 2 by HPLC
对化合物2进行解析,由碳谱化学位移得季碳为C1'δ175.60,由HMBC谱可知氢谱中与C1'相关氢谱峰为δ3.87~3.84、δ4.11、δ1.95、δ1.80-1.74峰,结合其他图谱可确定C1、C2'、C3',并通过C3'的COSY效应确定C4'。H谱中位于低场的δ5.70、δ5.69为双键C3'、C4',δ5.25、δ4.95为异头碳C1'、C1'',通过物质结构的分析可知只有H3'会与C1'产生HMBC远程相关信号,依此可同时确定4个C的归属。由H1'、H1''可在HMBC谱中确定C4、C6,并结合COSY确定C2、C3、C5,由H3'、H4'、H1'可通过COSY确定H2'、H5'、H6'。III环各C可通过已确定的C1''和6''(因其余4个仲碳都已确定)通过COSY谱进行归属。表2为化合物2的13C NMR、1H NMR、1H1H-COSY和HMBC的核磁谱解析数据。
同理,由碳谱化学位移得季碳为δ179.09的C1',由HMBC谱可得氢谱中与C1'相关的H峰,结合其他图谱可归属C1、C2'、C3',通过HSQC得到相连H的化学位移,并通过H3'的COSY效应确定H4'。除侧链两个仲碳外,δ63.08为与羟基相连的仲碳C6'',δ47.69为与氨基相连的仲碳C6',在剩余3个仲碳中,从找到HNBC谱找到与异头碳上的Hδ5.179或Hδ5.185远程相关信号,可确定C3'和H1'(同时得到H1'')。结合其他二维谱确定C4'和C2。由H1'与H1''可在HMBC谱中确定C4、C6,并结合H4和H6的COSY信号确定同环的H2、H3、H5。由H3'、H4'和H1'结合各二维谱信息确定同环的H2'、H5'、H6'。III环各C、H可通过已确定的H1''和H6'',通过COSY谱显示的同环自旋体系信息进行归属。
化合物1和化合物2的结构极为类似,化合物2的核磁谱解析见表2。
经MS鉴定化合物1和化合物2的分子离子峰[M+H]+分别为m/z553.5和551.4。碎片离子峰基本一致,[Ⅰ+Ⅲ+H]+为m/z425.1、m/z425.2,[Ⅰ+H]+为m/z264.0、m/z264.1,[Ⅲ+H]+为m/z162.9、m/z163.0。
经核磁和质谱分析,化合物1为3-氨基-3-脱氧-α-D-吡喃葡糖基-(1→6)-[2,3,4,6-四脱氧-2,6-二氨基-α-D-赤式-己吡喃糖基-(1→4)]-1-N-[(2S)-4-氨基-2-羟基-1-氧丁基]-2-脱氧-D-链霉胺,化合物2为3-氨基-3-脱氧-α-D-吡喃葡糖基-(1→6)-[2,3,4,6-四脱氧-2,6 -二氨基-α-D-赤式-3-烯-己吡喃糖基-(1→4)]-1-N-[(2S)-4-氨基-2-羟基-1-氧丁基]-2-脱氧-D-链霉胺。化合物1和化合物2属于2-脱氧-D-链霉胺双取代衍生物,其结构差异仅是己吡喃糖基上单键和烯键。图2为化合物1和化合物2的化学结构。
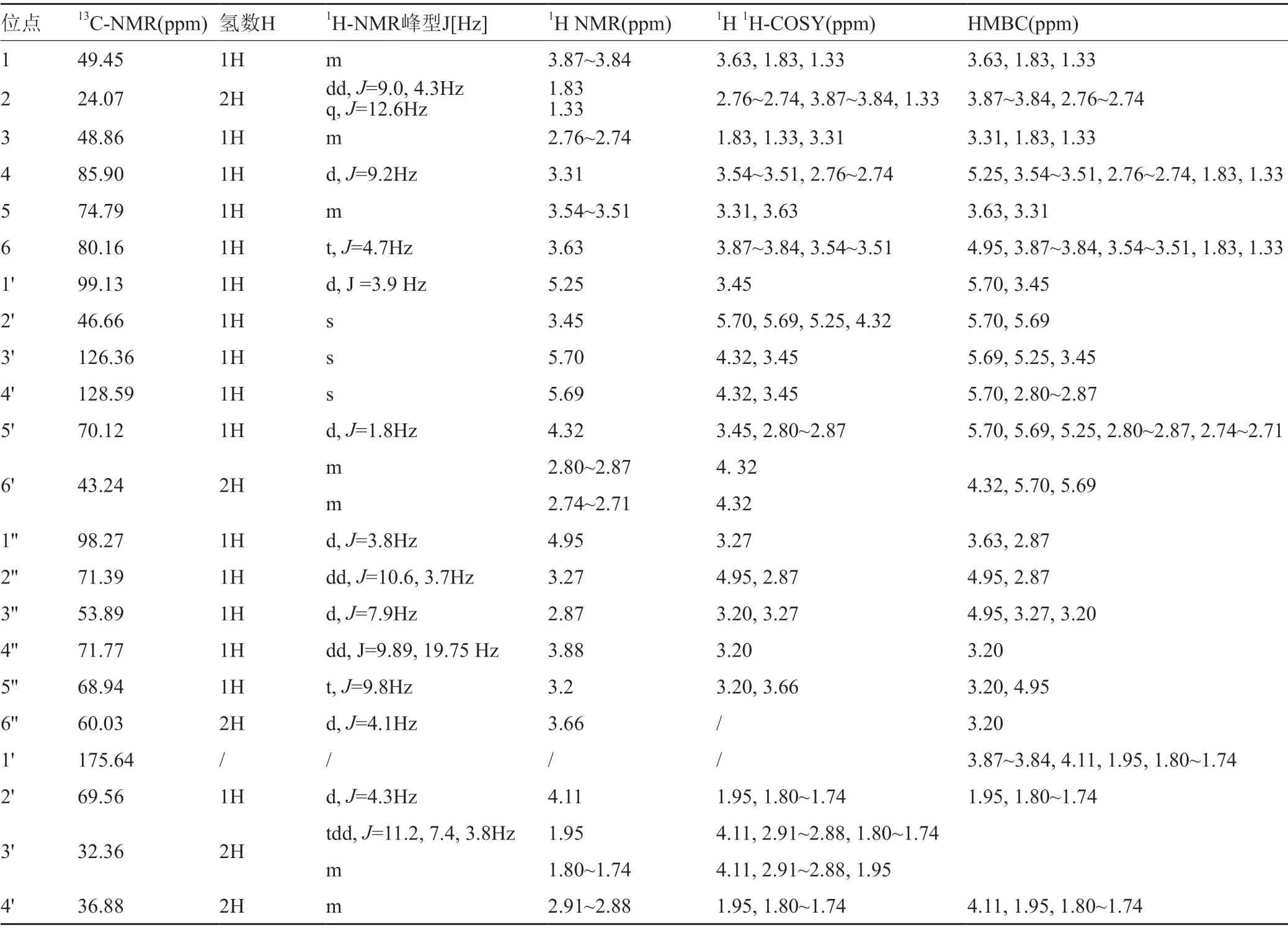
表2 化合物2的核磁谱解析Tab.2 NMR analysis of compound 2
2.3 内标物、定量峰的选择和含量测定
对分别加入内标物马来酸、甲酸钠的化合物1和化合物2进行1H NMR谱图分析(图3),马来酸易于识别的尖锐单峰δ6.23ppm归属为烯键氢,和化合物1的样品峰分离完全,互不干扰,因此马来酸可以作为化合物1的内标物。化合物1在δ5.05ppm、δ5.66ppm的位移是与氧代相连的碳C1'和C1''上的氢,是与其他信号分离的独立尖锐单峰,因此选择化合物1的δ5.05ppm、δ5.66ppm处的两个单峰信号为定量峰。同理,甲酸钠易于识别的尖锐单峰归属为δ8.36ppm,和化合物2的样品峰不发生重叠,因此甲酸钠可以作为化合物2的内标物。化合物2的δ2.00ppm位移是与C3'相连的-CH2,δ5.72ppm位移为烯键氢,这两组尖锐单峰与其他信号峰分离完全,因此选择该两组峰为化合物2的定量峰(由积分方法可知,核磁内标计算含量的计算方法不够详细,有无减少认为误差的措施)化合物1、2进行1H NMR内标法测定,样品重复测试6次,化合物1和化合物2的含量分别为64.98%±2.38%和75.38%±2.82%。

图2 化合物1和化合物2的化学结构Fig.2 Chemical structure of compound 1 and compound 2
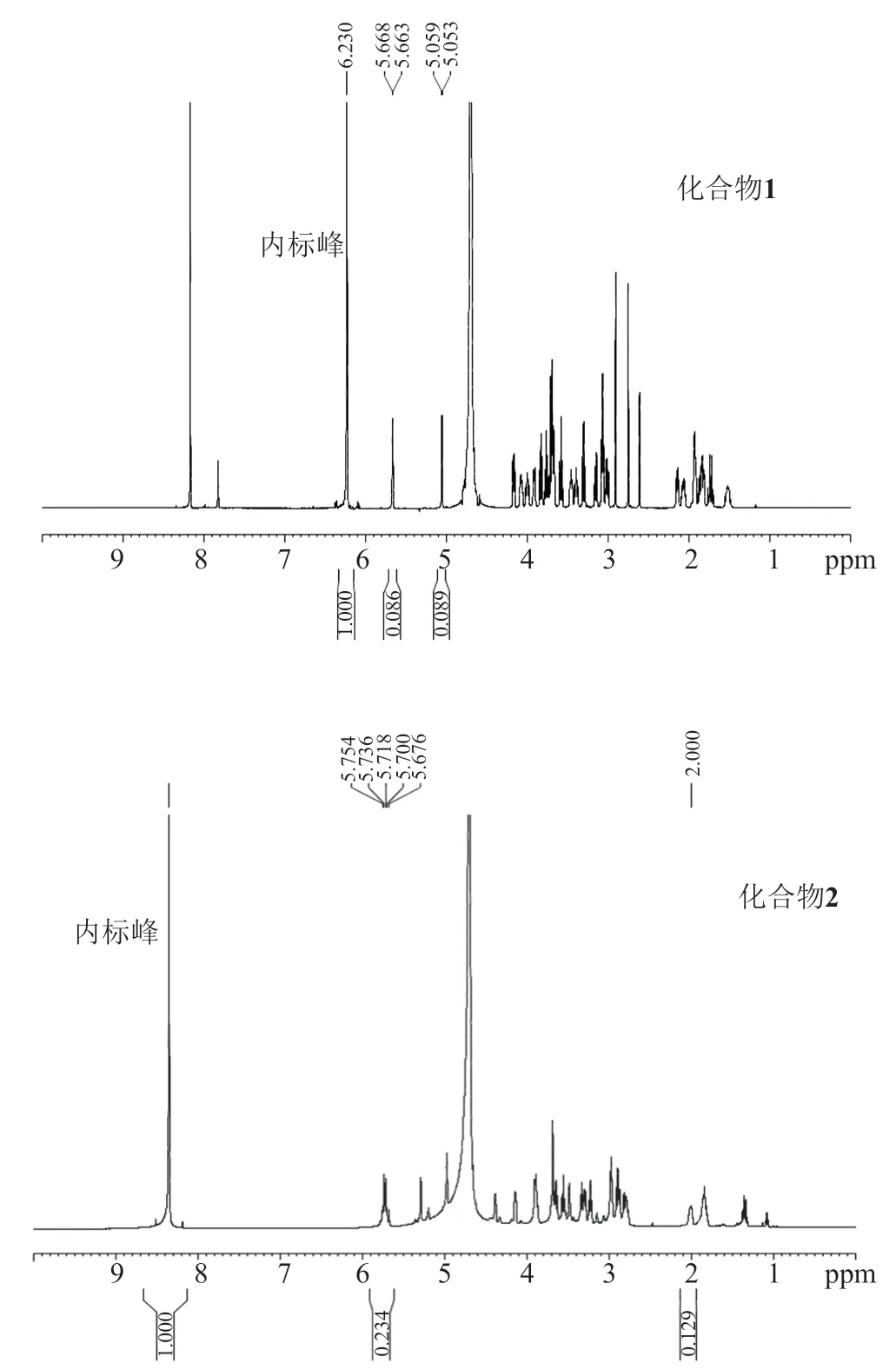
图3 化合物1、2和内标物的1H NMR相应峰Fig.3 1HNMR peak of compounds 1,2 and internal standards
3 讨论
化合物1和化合物2属于2-脱氧-D-链霉胺双取代衍生物,是第三代氨基糖苷类抗生素合成过程中的重要中间体。这两个化合物是结构非常接近的类似物,但分子结构中己吡喃糖基上单键和烯键的差异,使化合物的极性发生改变,因此无紫外吸收的化合物1和化合物2在邻苯二甲醛柱后衍生化HPLC分析中有较好的区分度,但无法购买商业化标准品是HPLC法定量测定的障碍。化合物1和化合物2的HPLC色谱纯度均大于95%,但由于样品易吸水、成盐性和杂质等干扰,色谱纯度并不能真正反映化合物1和2的浓度,需要提供足够量的样品以测定样品的水分及炽灼残渣。qNMR具有样品制备简单、无需标准品、准确、专一性高和不破坏样品等优点,是定量测定研发过程中样品量少且无商业化标准品化合物的有效方法。在qNMR分析中,弛豫延迟时间、激发脉角度、采样次数是影响定量的主要参数[9-10]。激发脉冲角度越小需要的弛豫延迟时间越短。本实验经过比较,在保证基线平直和高信噪比的条件下,激发脉冲角度30°、弛豫延迟时间20s、扫描次数选择16次时,样品和内标物马来酸、甲酸钠的定量峰积分可以满足准确度的要求。D2O对样品和内标物有较好的溶解性,且不产生干扰,因此选择D2O作为qNMR的溶剂。化合物1和化合物2的定量峰选择峰形较好,周围没有干扰的峰。内标物马来酸δ6.23ppm的烯键氢峰、甲酸钠δ8.36ppm的氢峰与所测样品的定量峰分离良好,且不产生干扰,因此选择马来酸和甲酸钠作为内标物。
越来越多的研究报道了qNMR测定的化合物浓度与质量平衡法浓度高度一致。qNMR法可以克服HPLC法缺少标准品带来的困扰,定性分析和定量分析可同步完成,方法简单、准确,为氨基糖苷类新药合成的过程检测和质量控制提供有效的技术手段。
猜你喜欢
中国药业(2022年22期)2022-12-08
现代仪器与医疗(2022年4期)2022-10-08
农业工程学报(2022年6期)2022-06-27
浙江化工(2022年1期)2022-02-19
四川大学学报(自然科学版)(2022年1期)2022-02-10
科学导报(2022年4期)2022-01-26
口腔护理用品工业(2021年4期)2021-11-02
蚌埠医学院学报(2020年11期)2020-12-17
中国科技纵横(2019年23期)2019-02-14
百姓生活(2016年6期)2016-06-22
