
氨苄西林晶体形貌模拟
2020-01-10 03:06尹秋响崔平平张美景侯宝红王静康
天津大学学报(自然科学与工程技术版) 2020年2期
尹秋响,赵 迅,崔平平,张美景,谢 闯,鲍 颖,侯宝红,王静康,周 玲
氨苄西林晶体形貌模拟
尹秋响1, 2,赵 迅1,崔平平1,张美景1, 2,谢 闯1, 2,鲍 颖1, 2,侯宝红1, 2,王静康1, 2,周 玲1
(1. 天津大学化工学院,天津 300072;2. 天津化学化工协同创新中心,天津 300072)

氨苄西林;螺旋生长模型;晶习;晶体生长
晶习指晶体的宏观形态,在晶体生长过程中,由于晶体的内部结构、与溶剂相互作用、添加剂等因素的不同,导致各晶面生长速度的差异,从而形成了不同的晶习.晶体的晶习不仅会影响产品本身的许多物理化学性质,如催化剂的反应活性,药物的生物利用度、流动性、黏附性等,在生产中还会对下游操作如过滤、干燥、储存等产生影响.
随着晶体产品的晶习越来越受到关注,为了节省晶习筛选实验耗费的大量时间和费用,借助理论模型模拟晶习发展迅速.目前用于晶习模拟的模型有基于晶胞几何结构的BFDH模型,基于周期键链理论的附着能(AE)模型,考虑溶剂作用的修正的AE模型,但这些方法都是静态方法,没有考虑晶体生长过程的具体动力学因素.Boerrigter等[1]开发了基于蒙特卡罗算法的模拟方法,该方法以晶体内分子间相互作用和溶剂的热力学参数为输入,被成功应用于扑热息痛、文拉法辛、阿斯巴甜等晶体的晶习模拟[2-4],但该方法只是用大量的随机取样模拟结晶的确定过程,大多时候只能用于验证结果的正确性.Doherty开发了螺旋生长模型,该模型在固体物理和表面化学的基础上,考虑了晶面螺旋的具体生长机理,被成功用于模拟洛伐他汀、奥氮平、α-甘氨酸等晶体的晶习[5-7].由于对共晶、溶剂化物的生长机理还缺乏深入了解,该模型的应用存在一定的局限性,但螺旋生长模型从动力学的角度模拟晶习,为理解晶体生长提供了新的认识.
氨苄西林是一种半合成青霉素,由改变天然青霉素母核6-APA的侧链得到,较第1代青霉素有更广泛的抗菌谱,常被用于治疗革兰氏阳性和革兰氏阴性菌引起的疾病.虽然已有较多对氨苄西林的晶体结构[8-10]、结晶过程[11-13]等方面的研究,但还鲜见有关其晶习的报道.为研究氨苄西林晶习形成的主要因素,本文利用CrystalExplorer[14]计算了氨苄西林晶体分子间作用力,在此基础上利用Material Studio分子模拟软件采用螺旋生长模型模拟了氨苄西林在水溶液中生长的晶习,并与BFDH模型、AE模型和蒙特卡罗方法模拟结果进行比较.更进一步从晶体生长单元间的相互作用出发,以螺旋生长机理为基础,从理论上解释了晶习的产生,为理解晶体生长提供了新思路,并与实验结果进行了对比.
1 计算模型
1.1 螺旋生长模型
1951年,Burton等[15]提出螺旋生长模型,解释了低过饱和度下的晶体生长与2D成核所需的高能垒之间的矛盾.图1(a)为晶体螺旋生长示意,晶面上的螺旋提供了晶体生长所需的台阶.图1(b)为晶体表面结构示意,平台是晶面上的完整部分,台阶是晶面上不完整层的边缘,溶液中的生长单元附着在台阶上的扭结点,使台阶向外生长.

图1 螺旋生长模型示意
晶面上边形的螺旋是由条螺旋边缘构成的.当台阶开始向外生长,下一条边缘随之出现,出现的边缘围绕螺旋的中心点生长,该过程不断重复并形成周期性的螺旋.晶面上每完成一个完整的螺旋,如图1(a)中1-2-3-4,晶面便出现新的一层,对应于一个晶面间距的高度,所以晶面的生长速率可以表示[16]为

式中:表示台阶高度,通常可以用晶面间距代替;表示晶面上形成周期性的一个完整螺旋所花费的 时间.
当台阶开始生长时往往需要先达到临界长度c,Chernov等[17]在溶菌酶的结晶实验中用在线原子力显微镜验证了这一现象并测量了台阶的临界长度. Voronkov[18]提出,台阶的生长速度可以表示为
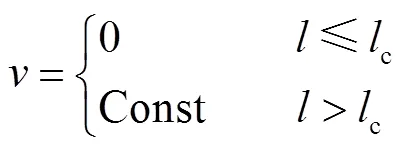
该方程表示当台阶的长度小于其临界长度时,其生长速度为0,只要其长度达到临界长度,台阶就会以一个恒定速度生长.
在达到临界长度前,当前螺旋边缘的延长是上一个台阶的生长导致的,所以螺旋生长总时间就是条螺旋边缘逐一生长至临界长度所花费的时间之和,可以表示[16]为
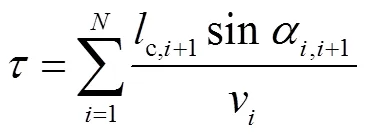
式中:v表示台阶的生长速度;,i+1表示前后两条螺旋边缘与+1的夹角;c,i+1表示台阶+1的临界长度.
对于台阶的临界长度,可以通过生长单元附着在台阶上的系统自由能变化计算.尽管这一过程对体积自由能的降低是有利的,但每个台阶的末端都存在新暴露的扭结面,增加了系统的表面自由能.Lovette等[19]提出,该过程的系统自由能变化可以表示为
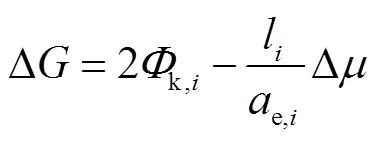
式中:k,i表示形成台阶末端扭结面所需的能量;e,i表示台阶上生长单元之间的平均距离.该过程前后系统自由能不变时对应的台阶长度即临界长度,可以表示为

最后,为计算晶面相对生长速度,还需要确定台阶生长速度v.螺型生长模型中,台阶生长速度v是台阶上的扭结点密度ρ、扭结点处生长单元的净附着率u和生长单元附着在扭结点时台阶生长的距离p,i的函数,可以表示为

1.1.1 净附着率
生长单元的净附着率u是扭结点处吸附速率+和脱附速率k-的函数,如果台阶上存在多种生长单元,则净附着率应是所有类型扭结点的平衡速率.图2表示对于有多种扭结点的台阶,其生长是生长单元有序进入台阶上不同扭结点的过程,该过程的吸附速率是恒定的,与发生吸附的扭结点位置无关,只取决于过饱和度;而脱附速率取决于生长单元在扭结点处与晶体的相互作用.根据台阶上生长单元与晶体的不同相互作用,本文研究的氨苄西林无水物在各晶面的每条台阶边缘上都有两种生长单元,故平衡状态下台阶生长的动力学过程可以表示为

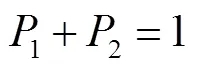

式中:P表示扭结点处为生长单元的概率;表示台阶边缘上存在的扭结点种类.式(9)表示A类型扭结点生成速率与A类型扭结点消失速率的平衡.可求得扭结点的净附着率为
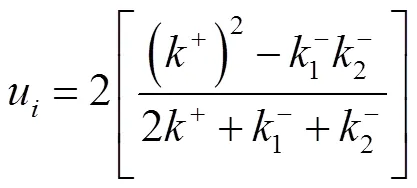
对于溶液结晶,生长单元在台阶边缘的吸附、脱附通量可以表示[20]为

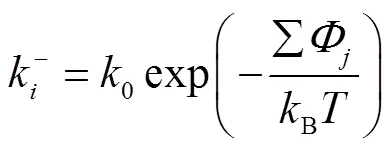

式中:∑Φ表示生长单元所有分子间作用力之和;∑Φ表示生长单元从扭结点脱附需要断裂的所有分子间作用力之和;0表示溶液中生长单元与晶面碰撞发生的频率.

图2 生长单元在台阶上的吸附脱附
1.1.2 扭结点密度
螺旋生长模型认为,扭结点是由于热力学波动导致的,所以扭结点密度与生成扭结点所需能量有关. Tilbury等[21]提出应依据各晶面扭结点的具体分布,考虑台阶上可能出现的所有类型扭结点,基于玻耳兹曼分布计算每种扭结点对扭结点密度的贡献.该模型取相邻生长单元的连接处为参考位点,考虑生成某种扭结点所需的能量贡献有:
(1)形成参考位点处左右两侧生长单元的平台面所需能量的1/2;
(2)形成参考位点处左右两侧生长单元的边缘面所需能量的1/2;
(3)形成参考位点处所有暴露在溶液中的扭结面所需的能量.
本文中氨苄西林的晶面上有两种可能的台阶结构,每种台阶结构都有两种扭结点:一种为单行的、两种扭结点交错排列构成的台阶,如图3(a)所示;另一种为单行A类型和单行B类型扭结点共同构成的台阶,如图3(b)所示.
对图3(a)表示的台阶,形成无缺陷台阶所需的能量可表示为
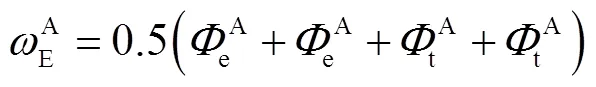
(15)
该台阶上有两种可能存在的单层扭结点(A,B)和两种可能存在的双层扭结点(AB,BA),形成每种扭结点所需的能量可分别表示为
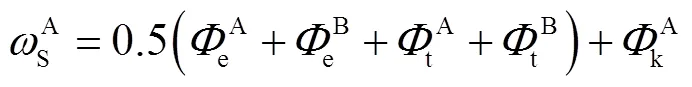
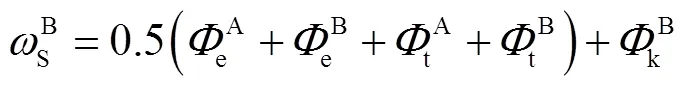

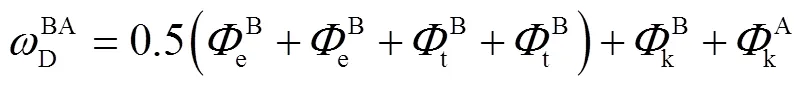
式中e、t、k分别表示形成扭结点边缘面、平台面、扭结面所需的能量,其值可表示为


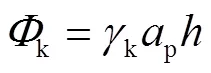
式中:e、t、k分别表示扭结点边缘面、平台面、扭结面的表面能;e、p、分别如图1(b)所示.
每种状态的概率可表示为

则扭结点密度可表示为
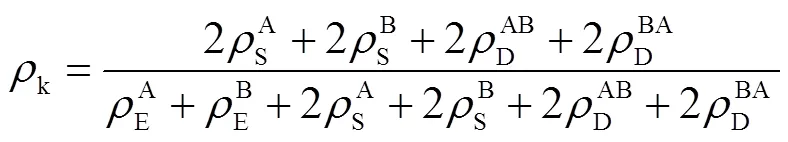
1.1.3 溶剂校正
Chernov[22]认为,晶面的表面能可以用与该晶面相交的生长单元间相互作用计算,固体-真空界面的表面能等于所有必须打破的生长单元间相互作用之和的1/2.但对于溶液结晶,溶液中的生长单元都处于溶剂化状态,而晶面上的生长单元是部分溶剂化的,两者的表面能都不能简单地用真空中模拟得到的分子间作用力计算,应考虑溶剂对分子间作用力的修正.依据组成的不同,本文将分子间作用力分为反映伦敦力的色散部分和反映极性力、氢键和静电作用的酸碱作用部分,分别进行校正.
纯溶剂的表面能可以通过Hansen溶解度参数计算.Beerbower[23]依据分子间作用力各成分对表面能的不同贡献,分别拟合了不同溶剂表面能与溶解度参数的关系式.
对酸、酚、胺和水,有


式中:下标d表示色散部分;下标a表示酸碱作用部分;m表示溶剂分子体积;d、p、h分别表示Hansen溶解度参数的色散、极性和氢键贡献.
晶体-溶液相界面的附着功可以用Girifalco-Good关系式对两相内聚能取几何平均计算,

则界面能可以表示为


本文所用25℃下水的溶解度参数[24]为d=15.5MPa1/2,p=16.0MPa1/2,h=42.3MPa1/2,分子体积m=18.0cm3/mol,还需用65℃下水的表面能对溶解度参数进行校正.
1.2 蒙特卡罗模拟
笔者应用螺旋生长机理,对各晶面的生长过程进行了蒙特卡罗模拟.模拟过程采用solid-on-solid(SOS)模型,该模型假设晶面上不存在悬空的生长单元,对于大多数的晶体生长过程,该近似是合理的.模型依据相互作用的坐标参数,确定晶面上生长单元与晶体的相互作用,而对边界处的生长单元,应用周期边界条件处理.不同于Kossel模型,氨苄西林的每个晶面上都有两种生长单元,故需要对每种生长单元分别建模.为更方便地描述各位点的相对位置,模型选取了晶面上较稳定的相交周期键链(PBC)作、、轴构建坐标系.
不同于平整晶面的生长,模型在初始晶面上引入了晶面间距高度的台阶以解释螺旋生长机理.如图4所示,图中表示位错面,、表示方向垂直于晶面向外、长度为晶面间距的Burgers矢量,模型假设穿过位错面的相互作用不变,则对于某特定的相互作用,面右侧生长单元与左侧原对应位置,高度差大于1的生长单元存在该相互作用.同时,为避免初始台阶消失,模型选取该晶面最稳定的PBC作为台阶延伸方向.

图4 位错示意
本文采用80×40的二维数组表示晶面,认为生长单元在晶面上可能发生吸附、脱附和表面扩散3种事件,每次模拟只能同时发生一种事件.吸附、脱附事件发生的概率可用式(11)和式(12)分别计算.对每个位点的表面扩散过程,模型认为生长单元只可能向相邻的、同平面上的8个位点扩散,但对于台阶附近的生长单元,可以发生跨越台阶的扩散.对于从脱附功为E的位点向脱附功为E的位点的扩散过程,其概率可表示[25]为

其中活化能DE可表示为

这种计算扩散概率的方式,既保证了基元反应的微观可逆性原则[26],又允许生长单元以较大的概率从脱附概率较高的位点向脱附概率较低的位点迁移,可以使模拟过程较快达到平衡,节省了模拟机时.
图5为模拟过程流程.对于作为输入的相互作用,还需要用氨苄西林在水中的溶解焓数据Ddiss=21.39kJ/mol进行标度[2-3,26-27].

首先根据输入参数对系统进行初始化,可以分别计算吸附事件的概率+、每个位点发生脱附事件的概率k-和每个位点各个方向上的扩散概率k,而每个位点发生扩散事件的概率k则为该位点各方向扩散概率中的最大值.然后通过生成随机数,判断本次模拟可能发生的事件,更新晶面上相关位点的高度,并使事件计数器加1.根据本次模拟发生的事件,分别更新相关位点的概率参数.晶面的相对生长速度可以根据模拟过程发生的总吸附次数att和总脱附次数det计算.

为保证模拟结果的准确性,模拟过程中每隔一定次数输出实时的att和det,并计算晶面的相对生长速度,取前后两次结果误差不大于5%作为系统稳定的标准.当过饱和度较低时,取决于不同晶面,系统总模拟次数有时需要1×106次才能达到平衡,但对于相对较高过饱和度,系统总模拟次数可以减少至3×105.

图5 蒙特卡罗模拟流程
2 实 验
2.1 实验试剂与仪器
试剂:实验所用氨苄西林(ampicillin,C16H19N3O4S)购自上海麦克林生化试剂有限公司;所用去离子水为实验室自制.
仪器:Julabo CF41恒温水浴槽,Julabo WB200-C搅拌器,Mettler Toledo ML204 分析天平,Hitachi TM3000扫描电镜,Rigaku D/MAX-2500粉末X射线衍射仪,奥特BK300光学显微镜.
2.2 氨苄西林的制备
氨苄西林主要以无水物和三水合物的形式存在.饱和水溶液中,在低于50℃结晶可得到三水合物,在60℃以上结晶得到无水物,不同温度下两种形式的溶解度差异导致其不同的溶解行为.
取50mL去离子水加入到结晶器中,升温至65℃,用2mol/L盐酸调至pH=2,在120r/min搅拌下加入1.8g氨苄西林,悬浊液在5min内溶清,用12mol/L氨水调至pH=5,10min内出现晶体,抽滤并在60℃下干燥
2.3 粉末X射线衍射表征
放射源Cu Ka,管压40kV,管流40mA,2衍射角5°~40°,扫描速率4.8°/min,步长0.02°.
3 结果与讨论
3.1 氨苄西林晶习模拟
3.1.1 螺旋生长模型
根据剑桥晶体结构数据库[8],氨苄西林的晶胞结构如图6(a)所示,晶格参数=1.240nm,=0.620nm,=1.2nm,=114.5°(AMCILL),属于单斜晶系P21空间群,晶胞中的氨苄西林以两性离子的形式存在.虽然单晶中重原子位置可以通过单晶X射线衍射得到,但晶体中氢原子位置无法准确估计,所以本文采用Material Studio中Dmol3模块的m11-L方法计算分子的静电势(ESP),并采用一致性价力场cvff[28]对晶体结构进行几何优化.
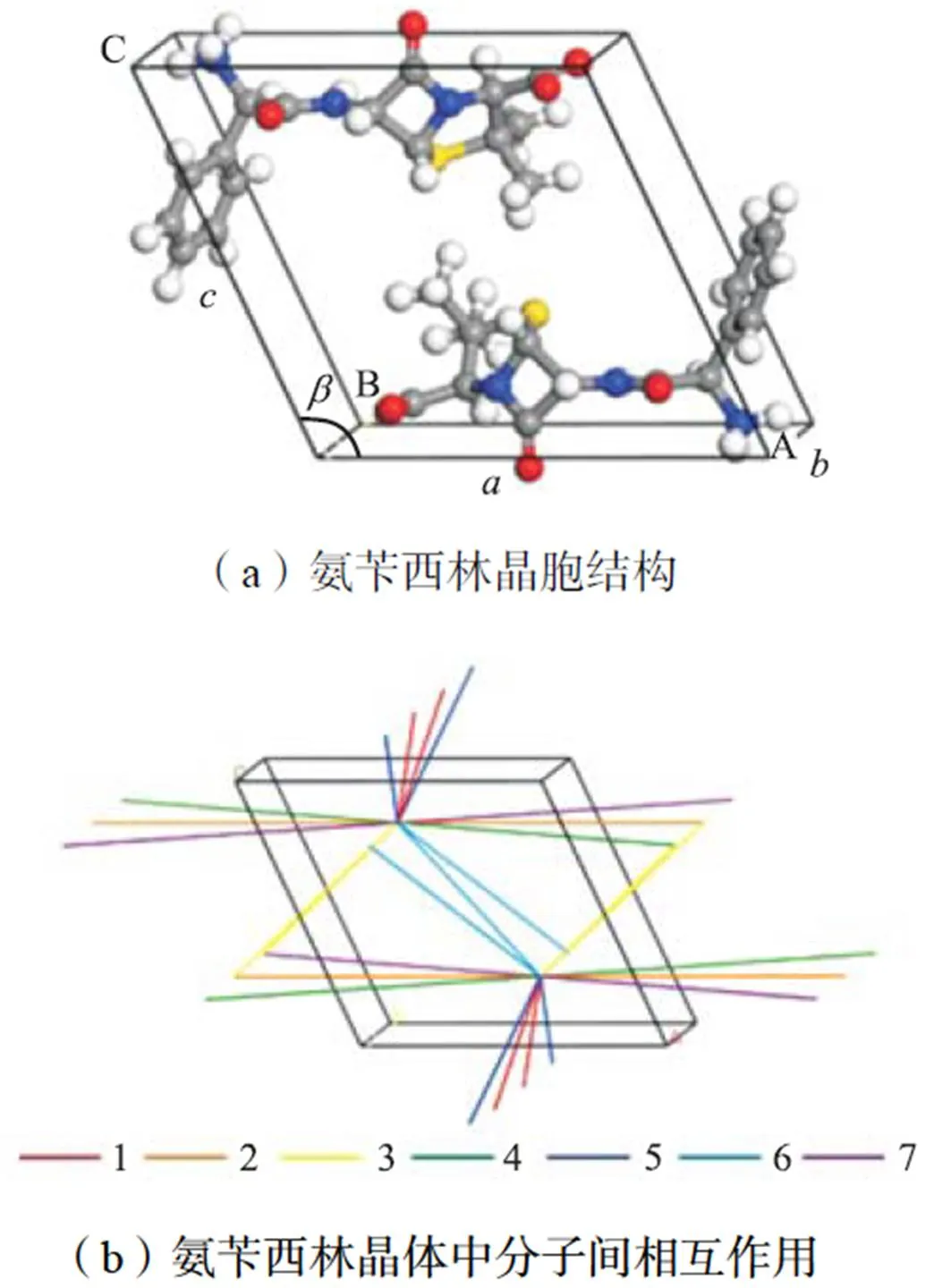
图6 氨苄西林晶体结构和分子间相互作用
表1为cvff力场优化前后的晶胞参数变化,可以看到,优化前后晶胞参数最大相对误差仅为2.82%,表明cvff力场优化后的氨苄西林晶胞适用于晶习模拟.
表1 晶胞参数文献值与优化结果比较

Tab.1 Cell parameters and optimization results
在模拟氨苄西林在水中的晶习过程中,最重要的是确定晶体各F面的相对生长速度.根据Hartman-Perdok理论,生长单元吸附在F面上释放的能量远小于其他晶面,该晶面生长速度最小,因此最容易在生长中暴露出来,是决定晶习的主要因素.晶体生长中的F面是包含2条及以上相交周期键链(PBC)的晶面,可以根据晶面上的相互作用分布确定晶体生长的F面.
本文通过CrystalExplorer中的Energy Framework计算得到晶体分子间相互作用,软件使用了校正的B3LYP-D2/6-31G(d,p)方法以得到较准确的作用力数据,计算得到的晶体中相互作用如图6(b)和表2所示.表2中相互作用的距离为两个分子重心间的距离,作用力大小为两个分子间所有相互作用的总和.这里特别指出的是,对于不同物质,当生长单元是二聚体、四聚体等时应该考虑生长单元进入晶格时需要克服的相互作用[29],本文模型物质的生长单元为氨苄西林分子,不涉及到多聚体,因此无需考虑多聚体内部的相互作用.
溶液-晶体相界面的形成本质上是克服晶体与晶体、溶剂与溶剂之间的分子间相互作用,再生成晶体-溶剂之间的相互作用形成相界面的过程,式(28)中的内聚能项解释了旧相互作用的断裂,附着能项解释了新作用力的生成.采用式(28)对模拟得到的晶体分子间相互作用进行校正,结果如表2所示.
表2 氨苄西林晶体中分子间相互作用

Tab.2 Intermolecular interactions of the ampicillin


图7 氨苄西林各晶面PBC结构

图8 晶面氨苄西林分子氢键分布
根据表3计算得到的各晶面相对生长速度,可以通过Material Studio的Morphology模块构建出氨苄西林的晶习,如图9所示.
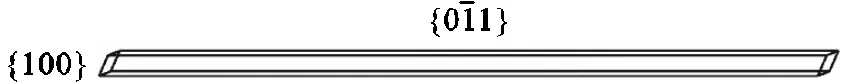
图9 螺旋生长模型模拟的晶习
表3 氨苄西林各晶面详细计算结果(=0.1)

Tab.3 Detailed results of the different faces of ampicillin(S=0.1)
3.1.2 BFDH模型和AE模型

图10 BFDH和AE模型模拟的晶习
表4 氨苄西林的晶面间距和附着能

Tab.4 Interplanar distance and attachment energy of ampicillin
3.1.3 蒙特卡罗方法模拟结果

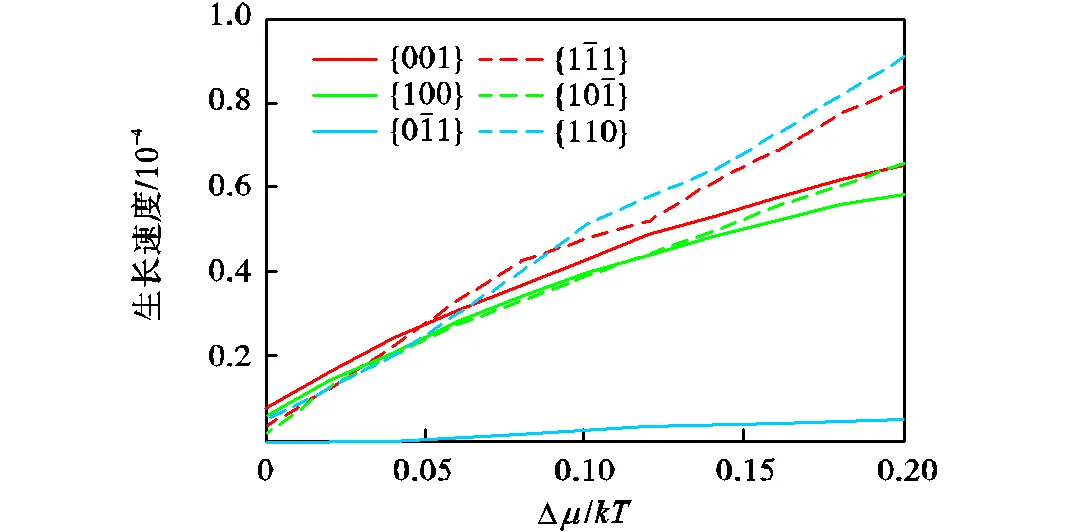
图11 蒙特卡罗方法模拟各晶面相对生长速度随Dm/kT, 变化

图12 蒙特卡罗方法模拟的晶习
3.2 氨苄西林实验结果


图13 氨苄西林XRD图

图14 实验得到的氨苄西林晶习
3.3 模拟结果讨论
比较BFDH模型、AE模型、蒙特卡罗方法和螺旋生长模型的模拟结果可以发现,螺旋生长模型模拟结果与水溶液中生长的氨苄西林晶习十分吻合,而BFDH模型和AE模型模拟的晶习均与实际晶习存在较大偏差.这可能是因为BFDH模型只考虑了晶体的物理几何性质,没有考虑到晶体中不同取向的相互作用.对于氨苄西林晶体,其分子间氢键和静电作用有强烈的方向性,同时溶液结晶过程中的溶剂化效应对晶体生长也有不可忽视的影响.而AE模型是在真空中模拟,一方面忽视了生长过程的动力学因素,另一方面也没有考虑到生长环境对晶习的影响,所以模拟结果也与实际晶习有一定差异.蒙特卡罗方法模拟结果在较高过饱和度下与实际晶习较为一致,但在低过饱和度下有一定的误差,这可能是由于输入的相互作用参数并不能简单地用溶解焓标度,同时受限于对晶体生长的理解,在晶面建模方面可能还需要进一步的改进.不同于AE模型,螺旋生长模型考虑了晶体生长过程的动力学因素,其速率控制步骤为生长单元进入扭结点的过程,在这个过程中前后台阶、前后扭结点、不同生长层之间的相互作用同样重要;且相比较于BFDH模型,还考虑了晶体-溶液相界面的溶剂化效应,因此该模型具有更实际的应用价值.实验证明,螺旋生长模型更适合于氨苄西林晶习.
4 结 语
[1] Boerrigter S X M,Josten G P H,van de Streek J,et al. MONTY:Monte Carlo crystal growth on any crystal structure in any crystallographic orientation;application to fats[J]. The Journal of Physical Chemistry A,2004,108(27):5894-5902.
[2] Boerrigter S X M,Cuppen H M,Ristic R I,et al. Explanation for the supersaturation-dependent morphology of monoclinic paracetamol[J]. Crystal Growth & Design,2002,2(5):357-361.
[3] Deij M A,van Eupen J,Meekes H,et al. Experimental and computational morphology of three polymorphs of the free base of Venlafaxine:A comparison of morphology prediction methods[J]. International Journal of Pharmaceutics,2008,353(1/2):113-123.
[4] Cuppen H M,Beurskens G,Kozuka S,et al. Crystal structure and growth behavior of aspartame form IA[J]. Crystal Growth & Design,2005,5(3):917-923.
[5] Kuvadia Z B,Doherty M F. Spiral growth model for faceted crystals of non-centrosymmetric organic molecules grown from solution[J]. Crystal Growth & Design,2011,11(7):2780-2802.
[6] Sun Y,Tilbury C J,Reutzel-Edens S M,et al. Modeling olanzapine solution growth morphologies[J]. Crystal Growth & Design,2018,18(2):905-911.
[7] Tilbury C J,Green D A,Marshall W J,et al. Predicting the effect of solvent on the crystal habit of small organic molecules[J]. Crystal Growth & Design,2016,16(5):2590-2604.
[8] Boles M O,Girven R J. The structures of ampicillin:A comparison of the anhydrate and trihydrate forms[J]. Acta Crystallographica Section B,1976,32(8):2279-2284.
[9] Shukla A,Khan E,Srivastava A,et al. A computational study on molecular structure,multiple interac-tions,chemical reactivity and molecular docking studies on 6 [D(-)α-amino-phenyl-acetamido] penicillanic acid (ampicillin)[J]. Molecular Simulation,2016,42(11):863-873.
[10] Larsen A S,Rantanen J,Johansson K E. Computational dehydration of crystalline hydrates using molecular dynamics simulations[J]. Journal of Pharmaceutical Sciences,2017,106(1):348-355.
[11] Ottens M,Lebreton B,Zomerdijk M,et al. Crystallization kinetics of ampicillin[J]. Industrial & Engineering Chemistry Research,2001,40(22):4821-4827.
[12] Ottens M,Lebreton B,Zomerdijk M,et al. Impurity effects on the crystallization kinetics of ampicillin[J]. Industrial & Engineering Chemistry Research,2004,43(24):7932-7938.
[13] Encarnación-Gómez L G,Bommarius A S,Rousseau R W. Crystallization kinetics of ampicillin using online monitoring tools and robust parameter estimation[J]. Industrial & Engineering Chemistry Research,2016,55(7):2153-2162.
[14] Turner M J,McKinnon J J,Wolff S K,et al. Crystal Explorer17[Z]. Australia:University of Western Australia,2007.
[15] Burton W K,Cabrera N,Frank F C. The growth of crystals and the equilibrium structure of their surfaces[J]. Phil Trans R Soc Lond A,1951,243(866):299-358.
[16] Snyder R C,Doherty M F. Predicting crystal growth by spiral motion[J]. Proceedings of the Royal Society of London A,2009,465(2104):1145-1171.
[17] Chernov A A,Rashkovich L N,Vekilov P G. Steps in solution growth:Dynamics of kinks,bunching and turbulence[J]. Journal of Crystal Growth,2005,275 (1/2):1-18.
[18] Voronkov V V. The movement of an elementary step by means of the formation of one-dimensional nuclei[J]. Sov Phys Cryst,1970,15:8-13.
[19] Lovette M A,Doherty M F. Reinterpreting edge energies calculated from crystal growth experiments[J]. Journal of Crystal Growth,2011,327(1):117-126.
[20] Hurle D T J. Handbook of Crystal Growth[M]. USA:Elsevier Science & Technology,1993.
[21] Tilbury C J,Joswiak M N,Peters B,et al. Modeling step velocities and edge surface structures during growth of non-centrosymmetric crystals[J]. Crystal Growth & Design,2017,17(4):2066-2080.
[22] Chernov A A. Modern Crystallography Ⅲ:Crystal Growth[M]. USA:Springer Science & Business Media,2012.
[23] Beerbower A. Surface free energy:A new relationship to bulk energies[J]. Journal of Colloid and Interface Science,1971,35(1):126-132.
[24] Hansen C M. Hansen Solubility Parameters:A User’s Handbook[M]. USA:CRC Press,2002.
[25] Xiao R F,Alexander J I D,Rosenberger F. Growth morphologies of crystal surfaces[J]. Physical Review A,1991,43(6):2977.
[26] Shim H M,Kim H S,Koo K K. Molecular modeling on supersaturation-dependent growth habit of 1,1-diamino-2,2-dinitroethylene[J]. Crystal Growth & Design,2015,15(4):1833-1842.
[27] Shim H M,Koo K K. Prediction of growth habit of β-cyclotetramethylene-tetranitramine crystals by the first-principles models[J]. Crystal Growth & Design,2015,15(8):3983-3991.
[28] Dauber-Osguthorpe P,Roberts V A,Osguthorpe D J,et al. Structure and energetics of ligand binding to proteins:Escherichia coli dihydrofolate reductase-trimethoprim,a drug-receptor system[J]. Proteins:Structure,Function,and Bioinformatics,1988,4(1):31-47.
[29] Hartman P,Perdok W G. On the relations between structure and morphology of crystals. I[J]. Acta Crystallographica,1955,8(1):49-52.
[30] Zhu H,Grant D J W. Influence of water activity in organic solvent+water mixtures on the nature of the crystallizing drug phase. 2. Ampicillin[J]. International Journal of Pharmaceutics,1996,139(1/2):33-43.
[31] Dauber-Osguthorpe P,Roberts V A,Osguthorpe D J,et al. Powder Diffraction:Theory and Practice[M]. England:Royal Society of Chemistry,2015.
Simulation of the Crystal Morphology of Ampicillin
Yin Qiuxiang1, 2,Zhao Xun1, Cui Pingping1,Zhang Meijing1, 2,Xie Chuang1, 2,Bao Ying1, 2,Hou Baohong1, 2,Wang Jingkang1, 2,Zhou Ling1
(1. School of Chemical Engineering and Technology,Tianjin University,Tianjin 300072,China;2. Collaborative Center of Chemistry and Chemical Engineering(Tianjin),Tianjin 300072,China)

ampicillin;spiral growth model;crystal habit;crystal growth
O782
A
0493-2137(2020)02-0169-11
10.11784/tdxbz201901032
2019-01-17;
2019-04-09.
尹秋响(1964— ),男,博士,教授.
尹秋响,qxyin@tju.edu.cn.
天津市应用基础及前沿技术研究计划资助项目(16JCZDJC32700).
Supported by the Tianjin Research Program of Application Foundation and Advanced Technology(No.16JCZDJC32700).
(责任编辑:田 军)
猜你喜欢
电镀与精饰(2022年11期)2022-11-15
检验医学(2022年2期)2022-03-14
人工晶体学报(2021年10期)2021-11-26
中西医结合心血管病电子杂志(2020年31期)2020-12-16
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
云南民族大学学报(自然科学版)(2015年4期)2015-11-14
中国新技术新产品(2014年4期)2014-03-12
大连医科大学学报(2010年3期)2010-01-25
