
中欧药物警戒制度的比较研究
2020-04-23 02:30胡歆雅梁玉清曾亚莉
中国合理用药探索 2020年1期
胡歆雅,梁玉清,曾亚莉,王 刚,梁 毅*
(1 中国药科大学国际医药商学院,南京 210000;2 科睿唯安,伦敦 SE18EZ)
药物警戒(pharmacovigilance,PV)一词的词根来源于希腊语Pharmakon(“药用物质”的意思)和拉丁语Vigilia(“保持警惕”的意思)。世界卫生组织(WHO)将其定义为“与发现、评估、理解和预防各种不良反应或与其他任何可能的药物问题有关的科学研究和活动[1]”。1961年,“沙利度胺海豹胎儿事件”的发生,反映出药品安全性监管方面的诸多问题,引起世界各国对动实验的可靠性以及药品上市后监测的重要性的关注与思考。1970年,WHO成立了药物监测中心(WHO Drug Monitoring Centre),1978年迁移至乌普萨拉,并更名为世界卫生组织国际药物监测合作中心,简称乌普萨拉监测中心(Uppsala Monitoring Centre,UMC)。自此,国际药品监管制度发生了巨大变化,药品安全性监测活动变得系统化、组织化和规范化。欧盟自2012年7月执行新版药物警戒法规,至今已形成了相当完备的药物警戒制度。而我国于2019年8月颁布新修订的《中华人民共和国药品管理法》(简称“新《药品管理法》”,下同),将药物警戒制度写入法律。目前来讲,我国药物警戒制度建设仍在探索阶段,需要借鉴先进的药物警戒理念与方法。因此,本文将中欧药物警戒制度进行比较,通过法律框架、监管范围、机构设置和数据库建设方面的比较分析,剖析我国药物警戒制度现状,为其进一步发展提出建议。
1 法律框架
1998年,中国开始建立药品不良反应监测与报告制度,鼓励企业自主上报药品不良反应。2004年3月原国家食品药品监督管理局和卫生部共同颁布《药品不良反应报告和监测管理办法》(局令第7号)。2011年,《药品不良反应报告和监测管理办法》(简称“81号令”,下同)发布,正式规定了药品生产企业、药品经营企业、医疗机构应报告药品不良反应以及药品监管部门在药品不良反应监管中所承担的责任和义务,详细说明了药品不良反应报告、定期安全性更新报告(PSUR)和重点监测的要求[2]。2017年,中国正式加入人用药品注册技术要求国际协调会议(ICH),开始逐步适用ICH指导原则,药物不良反应的报告和电子传输开始与国际接轨。2018年,《关于药品上市许可持有人直接报告不良反应事宜的公告》(简称“66号公告”,下同)正式发布,明确药品上市许可持有人(简称“持有人”,下同)建立药品不良反应监测体系、及时报告药品不良反应、加强不良反应监测数据的分析评价及采取有效的风险控制措施的主体责任,进一步完善了我国药品不良反应监测制度。2019年,新《药品管理法》正式将药物警戒制度立法,规定“国家建立药物警戒制度,对药品不良反应及其他与用药有关的有害反应进行监测、识别、评估和控制”,我国对药物警戒工作的重视程度进一步提升,药物警戒制度的建设与发展开始进入新篇章。中国药物警戒制度相关的主要文件见表1。
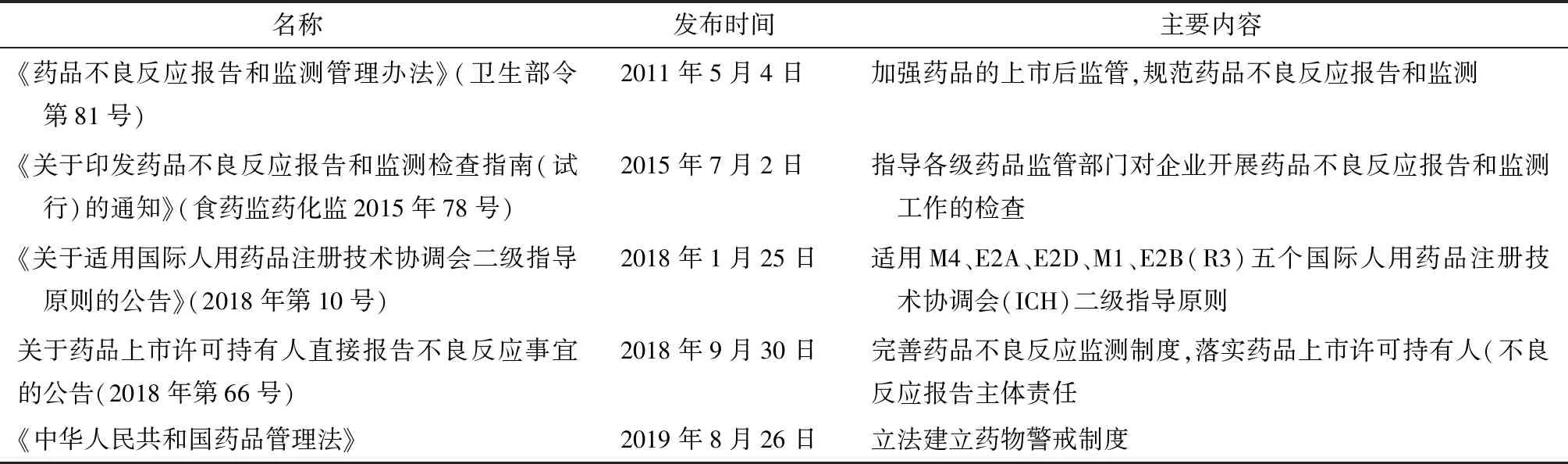
表1 中国药物警戒制度相关的主要文件
资料来源:科睿唯安Cortellis RI全球药政法规情报数据库
1965年1月,欧盟理事会颁布第65/65号(EEC)指令,标志欧盟药事立法的开始。2001年,欧盟颁布第2001/83号(EC)指令,统一了人用药品相关的规定,专门设置第9章规定药物警戒的总体要求。2010年通过的第2010/84号(EU)指令和第1235/2010号(EU)法规,于2012年7月正式生效,修订后的药物警戒法规对欧盟药物警戒制度进行了实质性的更新,完善了关于药品上市后监测的法律体系,明确了药品主管机构和持有人的分工和责任[3]。2012年,欧盟颁布了第2012/26号(EU)指令和第1027/2012号(EU)法规,提高了药物警戒制度的执行要求,特别是对药品安全性问题的及时通知和评估。同年,为支持新法的实施,欧盟制定了《药物警戒实践指南》(Good Pharmacovigilance Practices,GVP),为药物警戒工作的开展提供了一整套的指南。欧盟药物警戒制度立法的主要文件见表2。GVP共包括16个模块(见表3),涵盖药物警戒的主要方面,目前已制定完成,其中模块编号Ⅺ、Ⅻ、和因有其他指导文件替代而保持无效状态[4]。

表2 欧盟药物警戒制度立法的主要文件
资料来源:科睿唯安Cortellis RI全球药政法规情报数据库
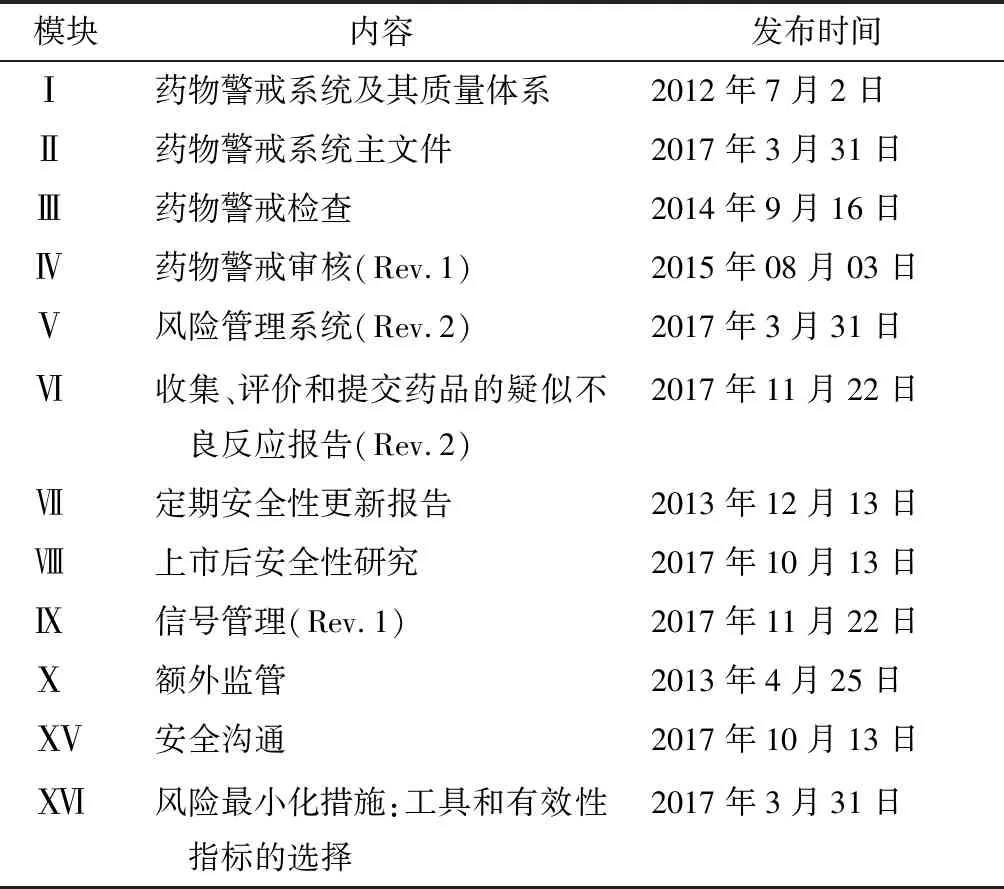
表3 欧盟GVP模块介绍
资料来源:科睿唯安Cortellis RI全球药政法规情报数据库
2 监管范围
第81号令规定了我国药品不良反应的报告范围,即合格药品在正常用法用量下出现的与用药目的无关的有害反应。2018年,66号公告的发布扩大了报告范围,规定按照可疑即报的原则,上报患者使用药品出现的与用药目的无关且无法排除与药品存在相关性的所有有害反应。因此,我国药物警戒的监管范围也随之扩大,包括药品上市后研究、风险管理计划、药品不良反应信息收集和反馈、监测数据定期分析评价等方面[5]。我国无菌药品警戒监测范围的扩大,一方面了扩大药品不良反应的收集规模,提升了药品安全性方面的监管质量;另一方面也让持有人从被动方转为主动方,通过建立自身的药物警戒体系,自发向药监部门报告,提高了药监部门处理药品风险的效率。
欧盟在2010年发布了第2010/84号(EU)指令,将药品不良反应的定义扩大为用药后出现的有害和非预期反应。这一举措不仅扩大了药品不良反应的上报范围,增加了药品不良反应的报告数量,也提高了药物警戒数据分析结果的质量。目前,欧盟药物警戒的监管范围涉及药品关键信息的收集、数据和信息的分析理解、维护公众健康的监管活动以及与利益相关方的沟通4个领域,包括风险管理计划、PSUR、患者和医护人员的不良反应报告、药物警戒系统主文件(PSMF)、药品信号检测等一系列行为[6]。这种严密的药品监测,能够让主管部门及时干预并尽可能减少与使用药物相关的不良反应或潜在风险的发生。
3 机构设置
我国药品不良反应监测机构是国家药品不良反应监测中心(NCADRM),该机构承担了国家药品不良反应报告和监测资料的收集、评价、反馈和上报,以及全国药品不良反应监测信息网络的建设和维护等职责。截至2011年末,我国已经建立34个省级药品不良反应监测中心和333个地市级药品不良反应监测中心,形成了从国家到县级的四级药品不良反应监测中心。由于我国国土辽阔,人口众多,医疗机构和药品生产企业遍布各地,因此这种层级分明,层层上传信息的机构设置能够适应我国的基本国情[7]。
欧盟的药物警戒工作机构是药物警戒风险评估委员会(PRAC)。2010年,欧洲药品管理局(European Medicines Agency, EMA)为加强欧洲药品的安全性监测,根据药物警戒立法正式成立了PRAC。PRAC是EMA负责评估及监测人用药物安全事务的科学委员会。它由成员国监管机构的药物安全专家、欧洲委员会提名的科学专家以及患者和医疗保健专业人员的代表组成[8],主要负责与不良反应风险相关的检测、评估、建议和沟通,在药品安全性评价方面发挥关键作用[9]。PRAC的独立设置既使自身成为药品监管系统中最先进的部分,也保证了欧洲药品的安全性和有效性。
4 数据库建设
国家药品不良反应监测系统,作为我国的药物警戒数据库,目前设有持有人报告、医疗机构/经营企业报告、监测机构管理3个入口。2018年,全国药品不良反应监测网络共收到149.9万份《药品不良反应/事件报告表》。1999~2018年累计收到1368万份《药品不良反应/事件报告表》,其中86.8%的报告来自医疗机构,来自药品生产企业的报告仅占5.1%。该系统目前仅持有人、医疗机构和监管机构能够查询使用;公众没有访问权限,因此无法获取药品不良反应相关的信息。目前,公众仅能通过《药品不良反应信息通报》获知我国上报的药品不良反应信息,但该通报发布时间无规律且自2010年后每年发布的期数明显减少[10]。
EudraVigilance,作为欧盟药物警戒数据库,是EMA用于监测所有已上市药品和临床试验中研究药品安全性的工具。根据第726/2004号(EU)法规,与EudraVigilance相关的活动主要包括:药品不良反应报告的收集与处理(包括医学文献检索到的报告)、欧盟所有批准药品信息数据库的维护和更新、风险信号的识别[11]。2017年11月12日,EMA发布新的EudraVigilance系统,增强了疑似不良反应报告和分析功能。2018年,EudraVigilance共收集201.6万份上市后疑似不良反应报告,拥有超过1450万份个例药品不良反应报告(ICSRs)。目前,欧洲地区公众可以访问EudraVigilance数据库,并查询和下载病例列表和病例报告表格。其他利益相关方,包括上市许可持有人、监管机构、医疗机构等都可以在一定权限内访问该数据库,获得各自需要的信息。
人工智能,作为近年新兴技术,针对来自药物警戒遇到的挑战,目前已经有相关解决方案。主要挑战包括:信息及时跟踪、管理和监控所有利益相关者、缩短上报监管机构时间、制定行动方案优先级、把控安全趋势、全面的审计准备工作等。
5 中欧药物警戒制度的比较
通过比较上述中国和欧盟的药物警戒制度现状,可以发现:第一,在法律框架方面,中国药物警戒相关的法律法规正处于快速更新的节奏中,特别是在66号公布发布之后;但相关法规过于碎片化,缺乏系统的药物警戒指导文件。第二,在监管范围方面,中国从之前的“狭义”药品不良反应扩大为使用药品出现的与用药目的无关且无法排除与药品存在相关性的所有有害反应,依然侧重于上市后监测,与欧盟覆盖药品全生命周期的药物警戒还存在差距。第三,在机构设置方面,基于中国国情的“地方模式”缺乏地方与地方的信息交流以及中央与地方的联系和有效沟通。第四,在数据库建设方面,我国药品不良反应监测系统,除了收集不良反应报告的基础功能,缺乏进一步的数据分析、预警功能和相应的风险防控,同时没有足够公开的访问权限设置。
6 对我国药物警戒制度建设的建议
6.1 建立系统的药物警戒制度框架
欧盟GVP为药物警戒的开展提供了一整套的指南,我国66号公告可以看做是建立该指南的探索,要求持有人建立药物警戒系统,设立专门的人员负责执行和管理。在此,建议我国参照欧盟GVP,结合现有法律法规,从药物警戒体系建设、药物警戒人员要求和培训、药物警戒文件管理、药物警戒审核等方面,制定一部既与国际接轨又符合中国实际的药物警戒实践指南。除了指导上市许可持有人之外,该指南还应明确药监部门及其下属部门的职责,明确医疗机构、药品经营企业和个人在药物警戒活动中的辅助作用。
6.2 形成覆盖药品全生命周期的药物警戒制度
药物警戒贯穿药品发展的始终,涵盖从药品上市前研究、上市后安全性监测及评价、直至最后的撤市或淘汰的整个药品生命周期。目前,国家药品监督管理局药品审评中心(CDE)负责药品上市前研究,包括药品注册、临床试验登记等;国家药品不良反应监测中心负责药品不良反应的收集和报告、上市后药品安全性监测及评价。也就是说,我国临床试验和上市后监测存在断层现象。一方面,建议我国制定关于药品上市前安全性研究的指南,开通药品风险的上报渠道,及时发现药品的潜在风险。另一方面,建议我国加强药品上市前研究和上市后监测的安全性方面的数据交流,实现数据互通;同时,适当调整机构职能,实现覆盖药品全生命周期的药物警戒制度。
6.3 完善我国药物警戒数据库
建议我国借鉴欧盟EudraVigilance数据库的先进功能,以及应用人工智能等先进技术,通过收集药品全生命周期的安全性信息,加强监测药品安全问题的能力,提高数据分析效率,优化不良反应报告程序。该数据库为药监部门、企业及其他利益相关者提供更有效的系统的同时,也要求解决药监部门地方与地方的信息交流以及中央与地方的联系和有效沟通问题。药品收集数据的全面性和完整性有助于药监部门早期发现并及时评估新的药物不良反应或已知药物不良反应的新变化(如频率或严重程度的改变),及时通知相关利益者,将药品安全性的风险降到最低。在权限访问方面,建议我国不良反应监测系统适当开放数据库的权限,供公众查询信息,而不是让药品安全数据和信息仅仅面向医疗机构、企业和监管部门。
猜你喜欢
中国合理用药探索(2022年1期)2022-11-26
现代仪器与医疗(2022年2期)2022-08-11
汽车工程师(2021年12期)2022-01-18
河北地质(2021年1期)2021-07-21
世界最新医学信息文摘(2021年12期)2021-06-09
建材发展导向(2021年23期)2021-03-08
科技传播(2019年22期)2020-01-14
小学生优秀作文(低年级)(2018年6期)2018-05-19
消费导刊(2017年20期)2018-01-03
求学·理科版(2016年3期)2016-03-23
