
黄芪生脉饮中多糖水解单糖指纹图谱的研究
2020-06-01 07:01侯晓蓉任立琴裘飞君2王鹏飞占扎君单伟光
浙江工业大学学报 2020年3期
侯晓蓉,任立琴,裘飞君2,王鹏飞,章 旋,占扎君,单伟光
(1.浙江工业大学 药学院,浙江 杭州 310014;2.浙江新光药业股份有限公司,浙江 绍兴 312000)
多糖(polysaccharide),由10个及以上的单糖分子通过脱水形成糖苷键连接而成的含醛基或羰基的多羟基聚合物[1],因其具有抗氧化[2]、抗肿瘤[3]、抗炎、抗凝血[4]等多种生物活性而受到了广泛的关注。据现代药理研究,黄芪含有多糖、胆碱、皂苷、氨基酸、甜菜碱、香豆素及黄酮等[5-6]。其中,黄芪多糖[7-9]可有效对抗异丙肾上腺素导致的心肌缺血大鼠的心电图ST-T段的异常变化,降低血浆乳酸脱氢酶、肌酸激酶活力,增加心肌组织过氧化氢酶和超氧物歧化酶的活力,并可减少丙二醛生成,减轻心肌细胞的形态变化,从而减少心肌缺血造成的损伤[10]。生脉饮[11]始见于唐·孙思邈《千金要方》,具有益气滋阴、敛汗生津之功,被历代医家广泛用于临床。因此,在生脉饮基础上加黄芪一味得黄芪生脉饮,黄芪生脉饮因其益气滋阴,养心补肺的疗效,越来越多地被用于气阴两虚、心悸气短的冠心病患者身上[12-13]。目前,对黄芪生脉饮中多糖的研究较少,关于其指纹图谱的建立未见报道,因此,笔者对黄芪生脉饮中的多糖进行水解及衍生化,并建立黄芪生脉饮多糖高效液相色谱指纹图谱,旨在较为全面地反映黄芪生脉饮中多糖水解单糖的种类与数量,进而对其质量进行整体描述和评价,对其质量控制具有重要意义。
1 材料与方法
1.1 材 料
1.1.1 仪 器
Agilent高效液相色谱仪(美国安捷伦有限公司,1260Ⅱ),XS205十万分之一天平(METTLER TOLEDO,XS205DU),电热恒温水浴锅(巩义市予华仪器有限责任公司,DF-101S),旋转蒸发仪(上海爱朗仪器有限公司,SB-1200),冷冻干燥剂(北京博伊康实验仪器有限公司,FD-1D-50),离心机(上海安亭仪器科学厂,TGL-16G)。
1.1.2 材料与试剂
材料:黄芪生脉饮由新光药业股份有限公司提供,批号分别为20140208,20140516,20140909,20150114,20150804,20151114,20160712,20170704,20171111,20180704。
试剂:三氟乙酸(TFA,分析纯,麦克林试剂有限公司),三氯甲烷(分析纯,上海凌峰化学试剂有限公司),甲醇(分析纯,江苏强盛功能化学股份有限公司),无水乙醇(分析纯,上海凌峰化学试剂有限公司),单糖对照品为D-甘露糖(Man)、D-核糖(Rib)、L-鼠李糖(Rha)、D-半乳糖(Gal)、D-木糖(Xyl)、D-阿拉伯糖(Ara)、L-岩藻糖(Fuc)、D-葡萄糖(Glu)、D-半乳糖醛酸(GalA)、D-葡萄糖醛酸(GluA)(分析纯,均购自国药集团化学试剂北京有限公司),1-苯基-3-甲基-5-吡唑啉酮(分析纯,上海麦克林生化科技有限公司),乙腈(色谱纯,美国天地试剂公司)。
1.2 实验方法
1.2.1 多糖样品水解
精确量取100 mL黄芪生脉饮口服液进行减压浓缩,得浸膏状黄芪生脉饮,加入20 mL蒸馏水进行复溶,再加入80 mL无水乙醇得到80%的黄芪生脉饮醇溶液,于4 ℃条件下静置过夜。静置后的醇溶液于高速离心机中4 ℃下3 000 r/min离心15 min,离心结束后弃去上清液,对沉淀部分进行减压浓缩以去除多余乙醇,冷冻干燥后得黄芪生脉饮总多糖样品。
精确称取黄芪生脉饮总多糖样品5.0 mg于耐压瓶中,加入2 mol/L三氟乙酸2.0 mL,120 ℃油浴水解6 h。冷却后向水解液中加入适量甲醇,旋蒸浓缩至干,以去除多余三氟乙酸,重复3 次,加入1 mL蒸馏水溶解后用于PMP(1-苯基-3甲基-5-吡唑啉酮)柱前衍生化。
1.2.2 PMP衍生化方法
1) 单糖标准品PMP衍生化
分别取400 μL单糖标准品溶液(2 mmol/L),加入400 μL 0.3 mol/L的NaOH溶液和400 μL 0.5 mol/L的PMP甲醇溶液,混合均匀,在70 ℃ 水浴锅中反应80 min,冷却至室温,加入500 μL 0.3 mol/L的HCl溶液进行中和,加蒸馏水300 μL,再加入等体积氯仿萃取3 次,取上层水相,用0.45 μm微孔膜过滤,待HPLC分析;取400 μL的混合单糖对照样品溶液(2 mmol/L),同法制备混合单糖样品PMP衍生物。
2) 样品衍生化
取400 μL多糖水解单糖溶液,对其进行衍生化,方法同上。
1.2.3 高效液相色谱分析条件
色谱条件为:流速0.8 mL/min,检测波长250 nm,柱温25 ℃,进样量10 μL,流动相为V(0.1 mol/L 磷酸缓冲盐)∶V(乙腈)=83∶17,等度洗脱。
1) 色谱柱的选择
对几种常用的色谱柱ZORBAX Eclipse XDB-C18(250 mm×4.6 mm, 5 μm),Shim-pack VP-ODS(250 mm×4.6 mm, 5 μm),ZORBAX Extend-C18(250 mm×4.6 mm, 5 μm),ZORBAX SB-C18(250 mm×4.6 mm, 5 μm),Ultimate XB-C18(250 mm×4.6 mm, 5 μm)以出峰时间及各峰之间分离度分别进行考察。
2) 流动相的选择
流动相中有机相种类及比例的选择:考察不同的乙腈比例下,各色谱峰的出峰时间及分离度情况,选择合适的等度洗脱比例;流动相中缓冲盐pH的选择:通过改变缓冲盐中Na2HPO4溶液与NaH2PO4溶液的配比,分别得到pH为6.6,6.8,7.0的3 个不同pH的缓冲溶液,观察不同pH下混合单糖对照品中各色谱峰出峰情况及各色谱峰之间分离度情况。
1.2.4 方法学考察
1) 仪器精密度考察
取衍生化反应后的同一批供试品溶液(批号S1),连续进样6 次,考察相对保留时间和相对峰面积的RSD值。
2) 重复性考察
取衍生化反应后的供试品溶液6 份(批号S1~S6)分析,考察相对保留时间和相对峰面积的RSD值。
3) 稳定性考察
取衍生化反应后的同一批供试品溶液(批号S1),分别于0,2,4,6,10,24 h进样,考察相对峰面积的RSD值。
1.2.5 样品测定
制备10批次黄芪生脉饮样品(S1~S10)的供试品溶液,得到其液相色谱图,采用“中药指纹图谱相似度评价系统”软件对样品中多糖水解单糖进行相似度的评价分析。
2 实验结果与分析
2.1 水解及衍生化条件优化
2.1.1 水解条件筛选
分别考察不同TFA浓度(1,2,4,6 mol/L)对样品水解程度的影响。按1.2.2节方法制备供试品溶液,水解所用供试品溶液各10 μL,按1.2.3节色谱条件进样分析。
现有的HPLC单糖组成分析方法一般先对样品进行水解处理,然后进行PMP柱前衍生化过程。水解使用的TFA浓度与样品水解程度有直接关系。由图1可知:随着水解所用TFA浓度的升高,共有峰的峰面积之和呈现先升高后降低的趋势。在TFA浓度低于2 mol/L时,色谱图中各主要色谱峰的共有峰面积之和较小,可能是由于样品水解不够完全;随着TFA浓度的升高,峰面积之和开始升高,当TFA浓度达到2 mol/L时,峰面积之和达到最大值,随后开始下降,最后基本没有变化,可能是因为TFA具有强酸性,浓度过高容易导致样品发生脱水反应,从而导致样品被破坏,水解后得到各单糖含量降低,各主要色谱峰的共有峰面积之和反而下降。因此水解样品使用TFA浓度过高或者过低都无法准确地对样品中的各单糖组分含量进行定量分析,最终确定水解时TFA浓度为2 mol/L。
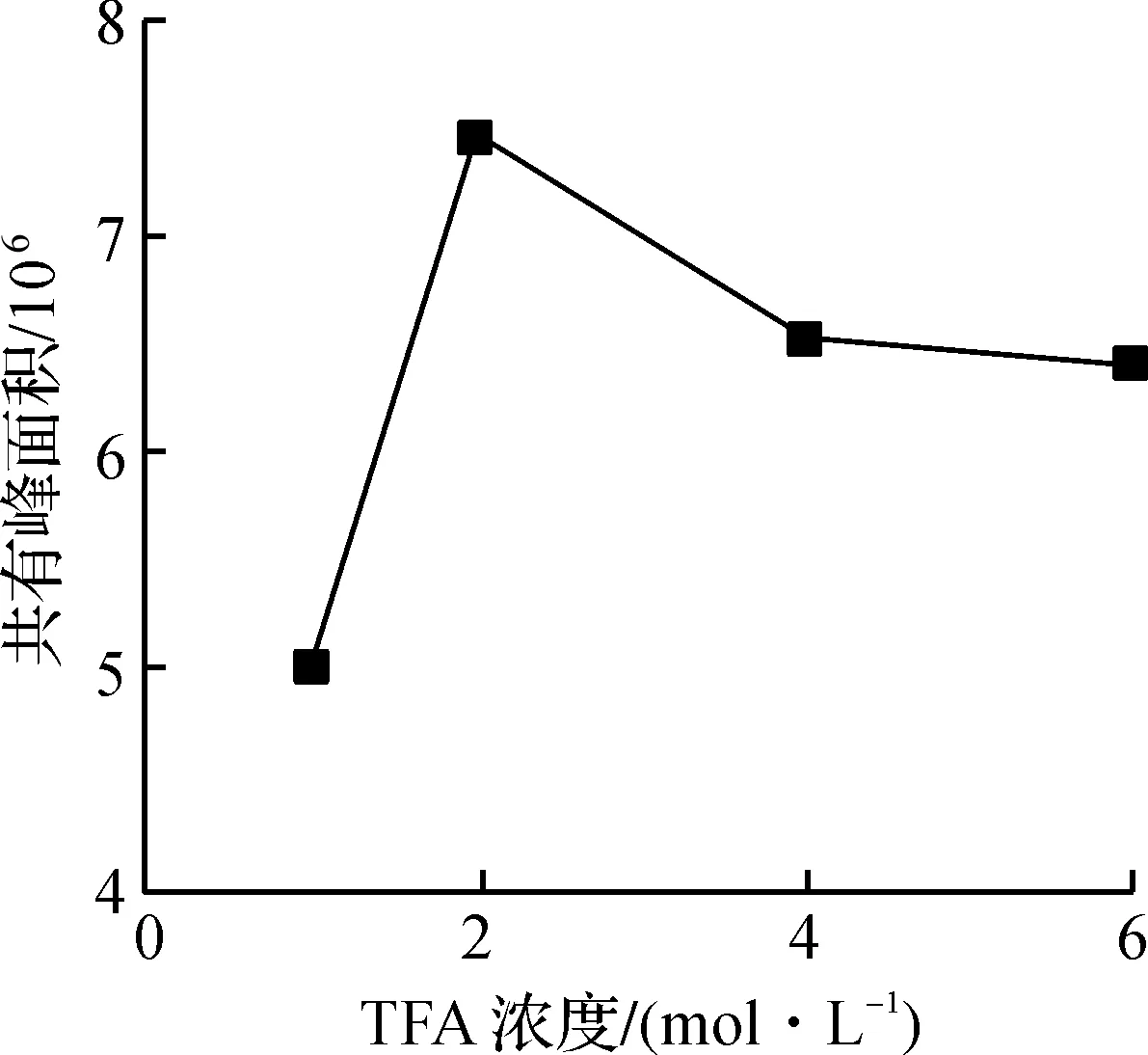
图1 不同TFA浓度水解对黄芪生脉饮多糖指纹图谱共有峰面积之和的影响(n=3)Fig.1 Effect of different TFA concentration on the sum of common peak areas of HPLC fingerprint of polysaccharide hydrolyzed monosaccharide of HSD(n=3)
2.1.2 PMP衍生化条件筛选
1) PMP衍生化时间的筛选
在衍生化反应的过程中,反应时间会对衍生化反应的程度产生较大影响。从图2可知:随着衍生化反应时间的延长,主要共有特征峰的峰面积之和呈先升高后降低的趋势。随着衍生化反应时间从30 min延长至100 min,共有峰面积和逐渐升高,当衍生化反应时间达到80 min时,各主要色谱峰的共有峰面积之和达到最大值,即可反应完全。当衍生化反应时间超过80 min后,随着反应时间的继续增加,各主要色谱峰的峰面积之和反而下降,说明在这种情况下,反应时间的延长并不能有利于衍生化反应的充分进行,可能是由于过长的衍生化反应时间导致了一些不明副产物的生成,从而导致各特征峰的共有峰面积之和下降。因此可确定衍生化最佳反应时间为80 min。相比于已经报道过的文献[4]中衍生化时间为100 min,本实验(80 min)减少了操作时间,提升了工作效率。

图2 不同衍生化时间对黄芪生脉饮指纹图谱共有峰面积之和的影响(n=3)Fig.2 Effect of different derivative time on the sum of common peak areas of HPLC fingerprint of polysaccharide hydrolyzed monosaccharide of HSD(n=3)
2) PMP衍生化试剂使用量的筛选
在衍生化反应的过程中,衍生化试剂PMP的使用量同样会对衍生化反应的程度产生一定影响。由图3可知:随着衍生化试剂使用量的增加,主要共有特征峰的峰面积之和呈现先升高后降低的趋势。当衍生化试剂的使用量高于400 μL时,随着PMP使用量的继续增加,各主要色谱峰的峰面积之和反而下降,说明在这种情况下,PMP用量的增大影响了衍生化反应的充分进行,可能是由于过量的衍生化试剂导致反应过程中副产物的生成,各特征峰的共有峰面积之和下降。因此可确定当总多糖样品为5.0 mg时,衍生化试剂PMP使用量为400 μL时衍生化反应效果较好。传统的衍生化反应过程中[14],通常使用过量的PMP,而强酸性溶液中含有碱性基团的PMP,由于PMP的质子化,很难在高效液相色谱分析之前被氯仿完全萃取。因此当总多糖样品为5.0 mg时,衍生化试剂PMP的使用量为400 μL不仅满足所有衍生物峰面积之和最大,且检测灵敏度最高,一定程度上减少了溶剂的使用量,符合现代绿色化学的理念。
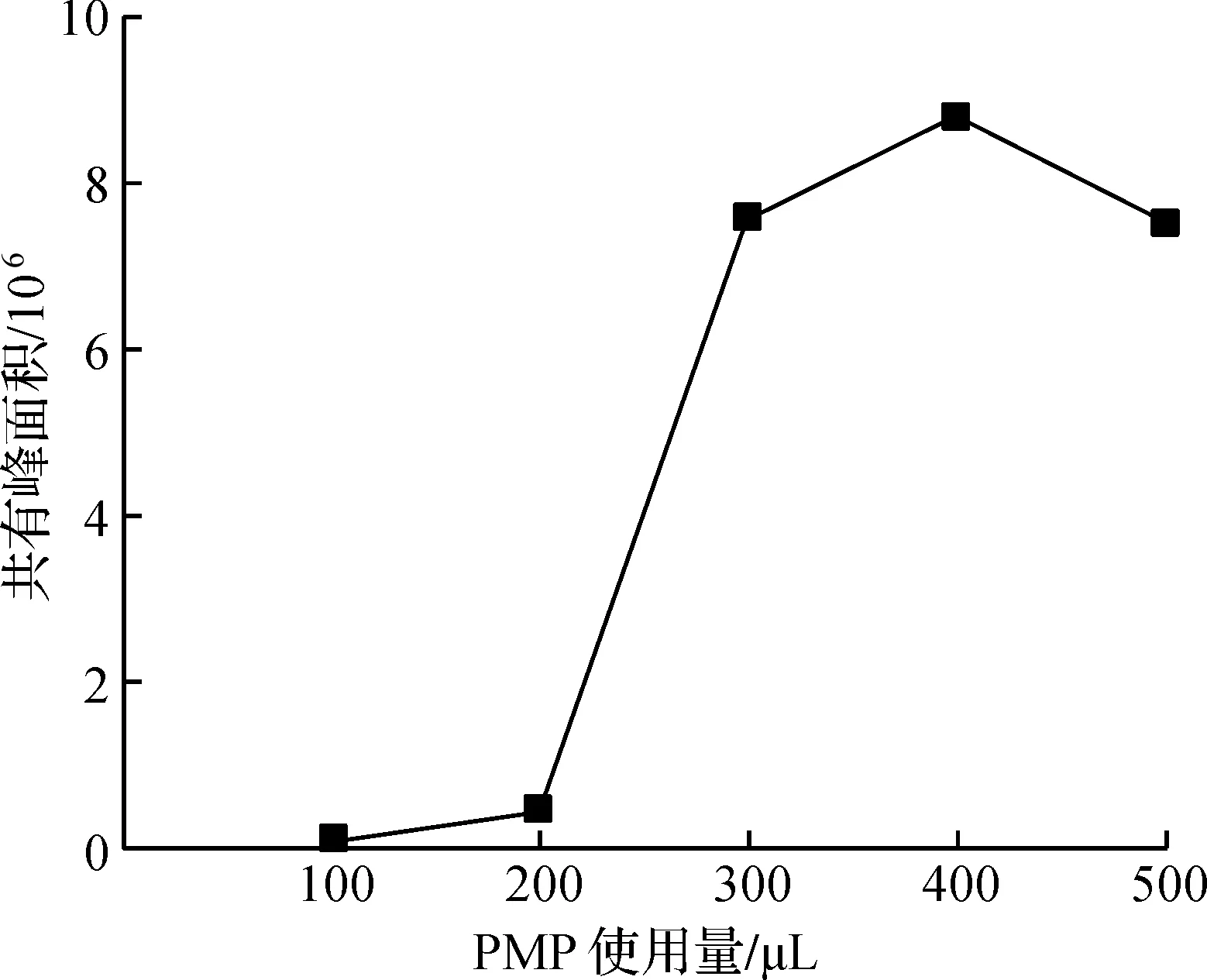
图3 衍生化试剂不同使用量对黄芪生脉饮指纹图谱共有峰面积之和的影响(n=3)Fig.3 Effect of different PMP(2.0 mol/L) amount on the sum of common peak areas of HPLC fingerprint of polysaccharide hydrolyzed monosaccharide of HSD(n=3)
2.2 色谱条件的优化
2.2.1 色谱柱的选择
比较ZORBAX Eclipse XDB-C18(250 mm×4.6 mm, 5 μm),Shim-pack VP-ODS(250 mm×4.6 mm, 5 μm),ZORBAX Extend-C18(250 mm×4.6 mm, 5 μm),ZORBAX SB-C18(250 mm×4.6 mm, 5 μm),Ultimate XB-C18(250 mm×4.6 mm, 5 μm)等5个型号的色谱柱,筛选得到Ultimate XB-C18色谱柱下的色谱图,不仅能够达到基线分离的要求,且各峰之间基线平稳,分离度均大于1.5,同等条件下其他色谱柱下色谱图或者达不到所有单糖成分均出峰或者分离度不好,故本实验分析项下的色谱柱均选用Ultimate XB-C18色谱柱。
2.2.2 流动相的优化
1) 流动相比例的优化
选择0.1 mol/L磷酸缓冲盐-乙腈作为流动相体系,通过改变有机相比例优化分析方法。当流动相中乙腈比例为19%时,部分峰之间无法达到基线分离;当流动相中乙腈比例为15%时,部分峰出现拖尾现象;当流动相中乙腈比例为17%时,各色谱峰之间能够达到基线完全分离,并且各峰形较好,整体符合HPLC指纹图谱要求。故本实验最终选用V(0.1 mol/L磷酸缓冲盐)∶V(乙腈)=83∶17作为流动相体系。
2) 流动相中缓冲盐pH的优化
色谱结果显示,当缓冲盐pH为6.6时,不能完全达到基线分离;当缓冲盐pH为6.8时,各色谱峰之间能够达到基线完全分离,且分离度大于1.5;当缓冲盐pH为7.0时,亦能达到基线分离且分离度大于1.5,保留时间延长,峰宽增加。故流动相中缓冲盐pH选用6.8为最终条件。
2.3 方法学考察
2.3.1 精密度实验
精密度实验结果见表1,2。结果表明样品中主要单糖如甘露糖、鼠李糖、葡萄糖等色谱峰的相对保留时间RSD值分别为0.60%,0.58%,0.54%,0.91%,1.03%,0.88%,0.95%,0.89%,0.90%,1.59%,均小于2.0%,相对峰面积RSD值分别为0.56%,0.84%,0.85%,0.33%,0.80%,0.65%,0.57%,0.92%,1.10%,均小于2.0%,表明该方法精密度良好,符合指纹图谱的测定要求。

表1 精密度实验:相对保留时间Table 1 The experiment of precision: relative retention time
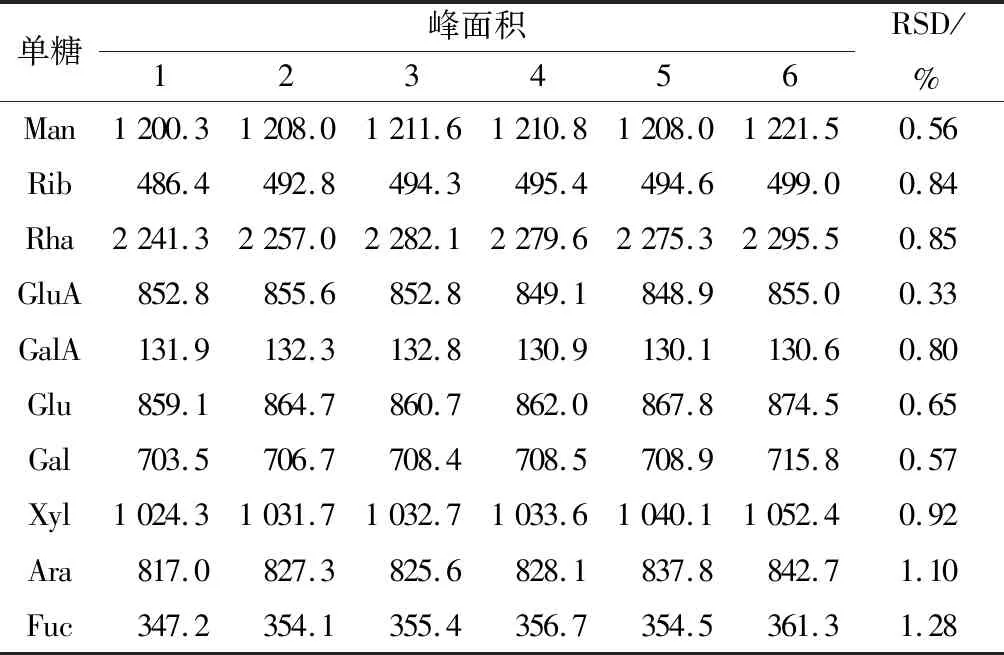
表2 精密度实验:峰面积Table 2 The experiment of precision: peak area
2.3.2 重复性实验
重复性实验结果见表3,4。结果表明样品中主要单糖如甘露糖、鼠李糖、葡萄糖等色谱峰的相对保留时间RSD值分别为0.71%,0.70%,0.68%,0.95%,0.97%,0.81%,0.83%,0.77%,0.75%,0.79%,均小于4.0%,峰面积RSD值分别为2.3%,3.7%,3.5%,0.7%,1.0%,3.7%,2.1%,2.1%,2.9%,2.3%,均小于4.0%,表明该方法重现性良好,符合指纹图谱的测定要求。

表3 重复性实验:相对保留时间Table 3 The experiment of repeatability: relative retention time

表4 重复性实验:峰面积Table 4 The experiment of repeatability: peak area
2.3.3 稳定性实验
稳定性实验结果见表5。结果表明样品中主要单糖如甘露糖、鼠李糖、葡萄糖等色谱峰的峰面积RSD值分别为2.30%,2.15%,1.81%,0.76%,0.92%,2.17%,2.51%,1.95%,2.45%,2.28%,均小于3.0%,表明该方法稳定性良好,符合指纹图谱的测定要求。

表5 稳定性实验:峰面积Table 5 The experiment of stability: peak area
2.4 指纹图谱的建立与分析
2.4.1 混合单糖对照品分析
精密吸取1.2.2节制备的混合对照品溶液,按照1.2.3节的色谱条件进行检测,得到色谱图如图4所示,混合单糖对照品10 个成分均能达到基线分离,且分离度均大于1.5,符合系统适应性要求。

1—甘露糖;2—核糖;3—鼠李糖;4—葡萄糖醛酸;5—半乳糖醛酸;6—葡萄糖;7—半乳糖;8—木糖;9—阿拉伯糖;10—岩藻糖。图4 10种单糖对照品的高效液相色谱图(n=3)Fig.4 HPLC chromatogram of ten reference monosaccharides (n=3)
2.4.2 共有峰及参照峰的确定
按照1.2.2节方法制备10批黄芪生脉饮的供试品溶液,按照1.2.3节色谱条件进行检测,色谱分析结果如图5所示。由图5可知:不同批次样品均具有基本一致的特征峰,共标示出10个主要共有特征峰。利用对照品保留时间以及色谱峰紫外光谱分析,鉴别出10个色谱峰,通过保留时间确定供试品溶液中1号峰为甘露糖,2号峰为核糖,3号峰为鼠李糖,4号峰为葡萄糖醛酸,5号峰为半乳糖醛酸,6号峰为葡萄糖,7号峰为半乳糖,8号峰为木糖,9号峰为阿拉伯糖,10号峰为岩藻糖(图5)。以峰10岩藻糖为参照峰分别计算各主要色谱峰的相对保留时间和相对峰面积,结果见表6,7。从单糖组成上可以看出:黄芪生脉饮样品中葡萄糖和阿拉伯糖的比例最大,其次是半乳糖、半乳糖醛酸、木糖、鼠李糖、岩藻糖、葡萄糖醛酸、核糖,其中以甘露糖的峰面积比例最小。
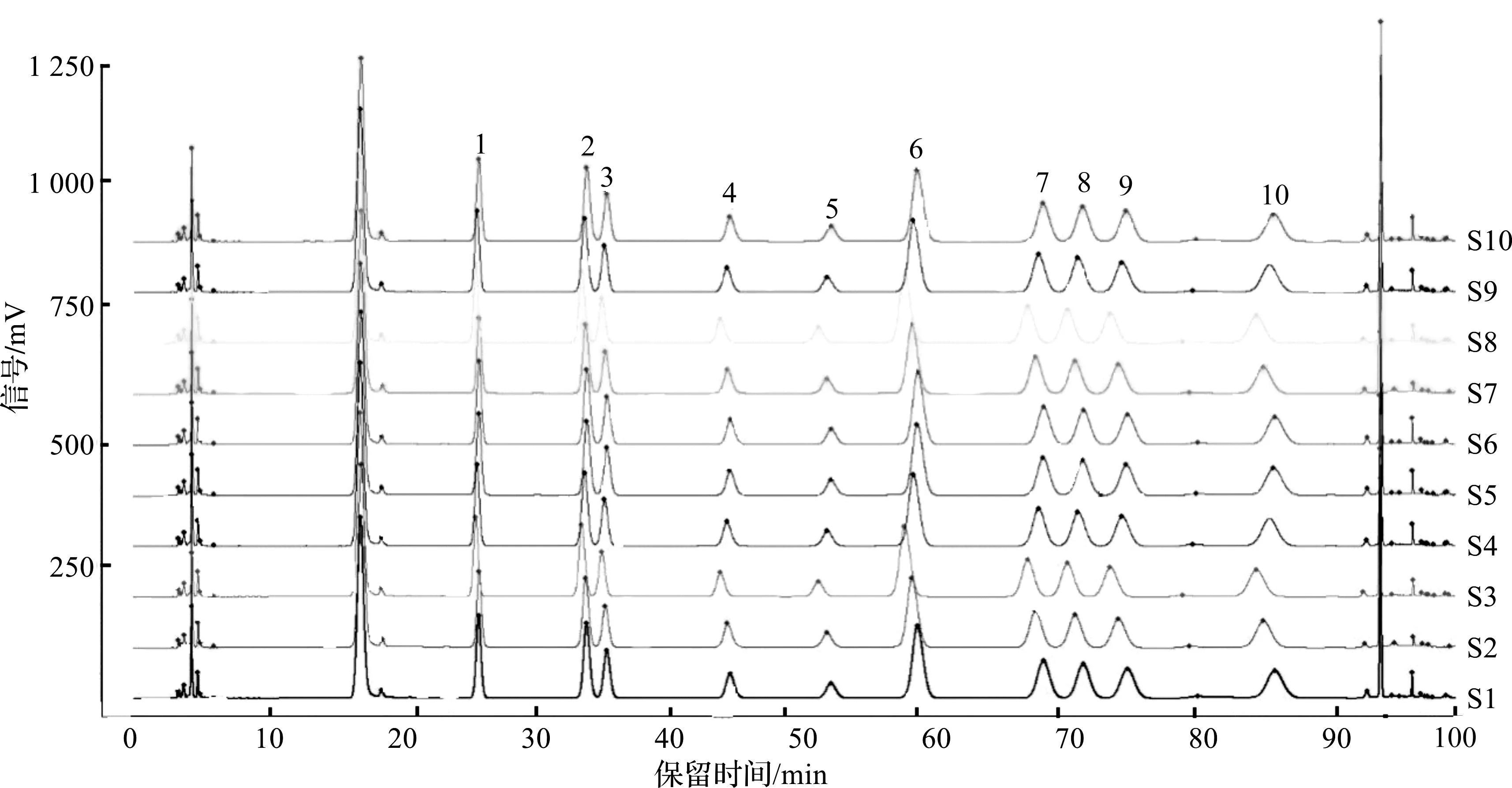
图5 10 批黄芪生脉饮水解物的高效液相色谱图Fig.5 HPLC chromatograms of HSD polysaccharide of ten batches samples
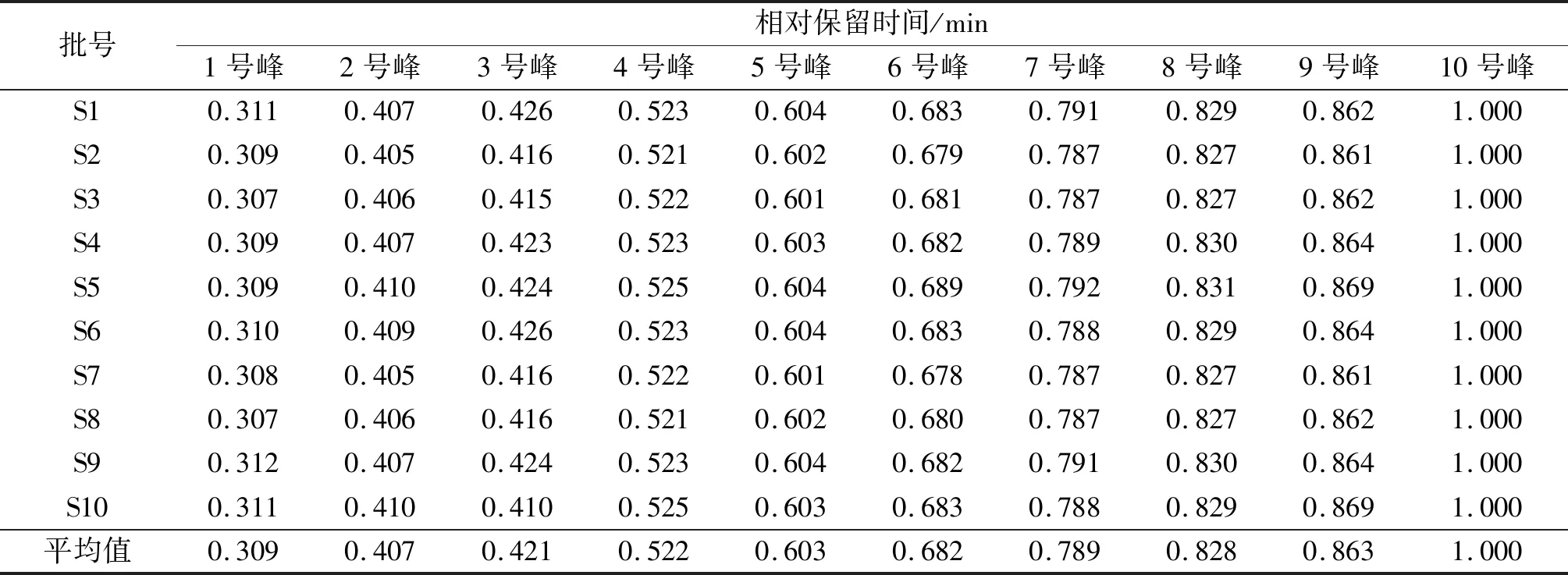
表6 10 批黄芪生脉饮水解物指纹图谱共有峰的相对保留时间(n=3)Table 6 Relativeretention time of common peaks for fingerprints of HSD of ten batches samples(n=3)

表7 10 批黄芪生脉饮水解物指纹图谱共有峰的相对峰面积Table 7 Relative peaks areas of common peaks for fingerprints of HSD of ten batches samples
2.4.3 相似度分析
采用计算机辅助相似度评价系统(国家药典委员会中药色谱指纹图谱评价系统软件2012.13072版)比较10批HSD供试品高效液相色谱指纹图谱全谱相似度,结果见表8:10批次HSD供试品高效液相色谱图谱全谱相似度均大于0.990,表明10 批黄芪生脉饮样品中含有的多糖成分具有很好的一致性,本方法可用于综合评价黄芪生脉饮的整体质量。

表8 10批HSD样品HPLC指纹图谱相似度分析结果Table 8 Similarity analysis results of HPLC fingerprint of HSD of ten batches samples
3 结 论
多糖相对分子量大、结构复杂、无紫外吸收,因此多糖色谱指纹图谱的建立比较复杂。选取从黄芪生脉饮中分离而来的多糖为实验对象,通过对多糖水解条件、PMP衍生化方法和液相色谱条件的摸索优化,建立了黄芪生脉饮多糖HPLC-UV指纹图谱法。研究结果表明:建立的HPLC指纹图谱方法具有较好的精密度、稳定性和重复性,可为黄芪生脉饮中多糖水解单糖的质量控制研究提供新方法。
猜你喜欢
基层中医药(2022年6期)2022-10-24
中国典型病例大全(2022年13期)2022-05-10
粉末冶金技术(2021年1期)2021-03-29
四川生理科学杂志(2020年1期)2020-03-20
中学化学(2019年3期)2019-07-08
教育教学论坛(2018年6期)2018-03-15
中国中药杂志(2017年20期)2017-11-11
科技资讯(2017年20期)2017-08-22
中学化学(2016年2期)2016-05-31
课程教育研究·下(2016年2期)2016-03-25
