
CO2 对酰基化合物引发2-吡咯烷酮聚合的影响
2020-06-03 12:58赵黎明邱永隽
功能高分子学报 2020年3期
陈 晨, 陈 涛,2, 赵黎明, 邱永隽
(华东理工大学1. 材料科学与工程学院,上海市先进聚合物材料重点实验室;2. 中国轻工业生物材料工程重点实验室;3. 生物工程学院,生物反应器工程国家重点实验室,发酵工业分离提取技术研发中心,上海 200237)
聚氨基丁酸(PGABA,又称聚酰胺4、聚丁内酰胺、聚2-吡咯烷酮)是一种生物基合成聚酰胺,单体2-吡咯烷酮(Py)源自γ-氨基丁酸[1],后者是一种可再生原料,可以通过大肠杆菌发酵生物质得到[2]。PGABA 的耐热性、断裂伸长率、韧性、弹性恢复与聚酰胺6 相似,吸湿性更优与棉花相似[3,4],并且主链结构接近天然蛋白,因而具有良好的生物降解性,可以在堆肥土壤、活性淤泥、活体内降解[5-8]。随着环境污染、石油资源短缺等问题日益严重,PGABA 作为一种性能优良的可降解生物基聚酰胺正得到越来越多的关注。但因其熔点与热分解温度接近,无法熔融加工,抑制了其商品化及规模化应用。目前解决这一问题的途径主要有两个:一是通过溶液静电纺丝制备PGABA 纤维膜,由于其良好的力学性能和生物相容性,PGABA 纤维膜在过滤和生物医用领域有较大的应用潜力[9];二是通过提高分子量、共聚[10,11]、端基改性[12]等方法来提高PGABA 的热稳定性。
传统的吡咯烷酮盐催化2-吡咯烷酮阴离子开环聚合制备PGABA 的方法聚合速率缓慢,产物分子量分布宽、分子量低、热稳定性差,极大地限制了PGABA 的工业化发展。此后,研究者们对聚合条件进行了一系列优化,研究表明在盐催化体系中加入酰基化合物做引发剂可以明显提高反应速率和产率,但是产物仍然存在分子量分布宽、分子量低等缺点[13,14]。在无引发剂的催化体系中直接通入CO2制备PGABA,其分子量分布较窄、分子量和热稳定性较高,但是与酰基化合物引发的聚合反应相比,此反应时间长、产率低[15]。迄今为止,对CO2在2-吡咯烷酮聚合中的作用有不同的观点。研究表明,酰基化合物引发剂和CO2均有清除链终止杂质,加快聚合速率的作用[16],CO2并非引发剂而是起到了促进剂的作用[17]。
本文在酰基化合物引发剂存在下将CO2引入Py 聚合体系共同促进聚合反应,详细探讨了催化剂和CO2用量对聚合结果的影响。

表 1 聚合反应投料比对产物的黏均分子量和产率的影响Table 1 Effects of reaction formulation on Mη and yield of products
1 实验部分
1.1 原料和试剂
生物基2-吡咯烷酮:实验室自制;碱A(氢氧化钠)、酰基化合物引发剂B(乙酰氯):分析纯,上海麦克林生物科技有限公司,直接使用;CO2:纯度>99%,上海申中气体有限公司,使用前用无水氯化钙和硅胶干燥。
1.2 PGABA 的合成
本文通过下述3 种方法制备PGABA。3 种反应方法投料配比及所得产物的产率、黏均分子量(Mη)见表1。
1.2.1 酰基化合物B 引发聚合 将一定量的2-吡咯烷酮置于三口烧瓶中,升温至90 ℃,加入适量的碱A 制备吡咯烷酮盐(催化剂),反应一定时间后移除小分子副产物,然后降温至40 ℃,加入计量的引发剂B,在60 ℃下反应30 min 后产物开始固化析出,24 h 后得到白色固体产物,分离后用水洗涤,抽滤后烘干至恒重。
1.2.2 无引发剂通入CO2进行聚合 在上述反应制备出吡咯烷酮盐后不加入引发剂,而通入一定量的CO2进行聚合,其他反应条件和后处理不变。
1.2.3 引发剂B 和CO2协同作用下的聚合 在1.2.1 节所述反应中,加入计量的引发剂B 的同时通入一定量CO2进行聚合,其他反应条件和后处理不变。
1.3 测试与表征
PGABA 易吸水,测试之前所有样品均干燥至恒重。采用德国Bruker Advanced 500 型核磁共振波谱仪测定产物的核磁共振氢谱(1H-NMR),溶剂为CDCl3-TFA(TFA:三氟乙酸)混合物(体积比为1/1);采用美国热电公司Nicolet5700 Fourier 型傅里叶转变红外光谱(FT-IR)仪对产物化学结构进行表征;采用黏度法测产物分子量,溶剂为甲酸,测试温度30 ℃,黏均分子量根据公式计算,其中:[η]为特性黏数,ηrel为相对黏度,由溶液流出时间与纯溶剂流出时间的比值决定[12];采用美国Waters 公司1515-2414 型凝胶渗透色谱(GPC)仪对产物的分子量分布进行测试,六氟异丙醇为流动相,流速为1.0 mL/min,以聚苯乙烯为标样的校正曲线校准;采用日本理学公司D/Max B 型X 射线衍射仪测量大角X 射线衍射(WAXD)谱图表征产物结晶形态;采用TA 公司Q2000 型差示扫描量热仪(DSC)测定产物热性能和结晶性能,升温速率10 ℃/min,测量温度范围为30~275 ℃;采用德国NETZSCH(耐驰)STA409PC 型热重分析(TG)仪表征产物热分解行为,氮气气氛,升温速率为10 ℃/min,测量温度范围为30~600 ℃。
2 结果与讨论
2.1 PGABA 的化学结构分析
图1 为PGABA 的1H-NMR 谱。其中PGABA-CO2为仅引入CO2进行聚合的产物,PGABA-2 为仅由引发剂B 引发聚合的产物,PGABA-3 为引发剂B 和CO2共同作用下的聚合的产物。由图可知,不同方法合成的PGABA 在化学位移2.0、2.6、3.5 处均出峰,且峰强比例大致为1∶1∶1,分别归属于PGABA 结构单元中2,3,4位的3 个亚甲基的氢原子,由此可以判断3 种方法均可成功合成PGABA。
图2 为上述3 种PGABA 样品的FT-IR 谱图。由图可知,不同方法合成的PGABA 的红外特征吸收峰没有明显的区别。所有PGABA 样品均在2 956 cm−1处出现C―H 伸缩振动峰;在3 297 cm−1和3 071 cm−1附近出现N―H 的游离吸收峰和明显的氢键吸收峰;1 649 cm−1和1 545 cm−1对应酰胺Ⅰ和酰胺Ⅱ的特征峰。FT-IR分析表明,这3 种方法均可合成PGABA,且PGABA 的羰基与亚氨基之间存在较强的氢键作用。

图 1 PGABA 的核磁共振氢谱Fig. 1 1H-NMR spectra of PGABA

图 2 PGABA 样品的红外光谱图Fig. 2 FT-IR spectra of PGABA
Galimberti 等[18]早前研究指出,PGABA 存在α 和γ 两种晶型,在一定条件下会发生晶型间的转变。图3为3 种方法合成的PGABA 的WAXD 谱图。由图可知,3 种聚合方法的产物均在2θ 为20.6°,24.4°处有2 个明显的聚酰胺类特征峰。由Jade 软件对谱图进行分析得到其面间距为0.431 nm,层间距为0.364 nm,分别对应PGABA α 晶型的200 晶面和020 晶面,即3 种方法合成的PGABA 在晶型上没有差别,都属于α晶型。
2.2 PGABA 的分子量及其产率
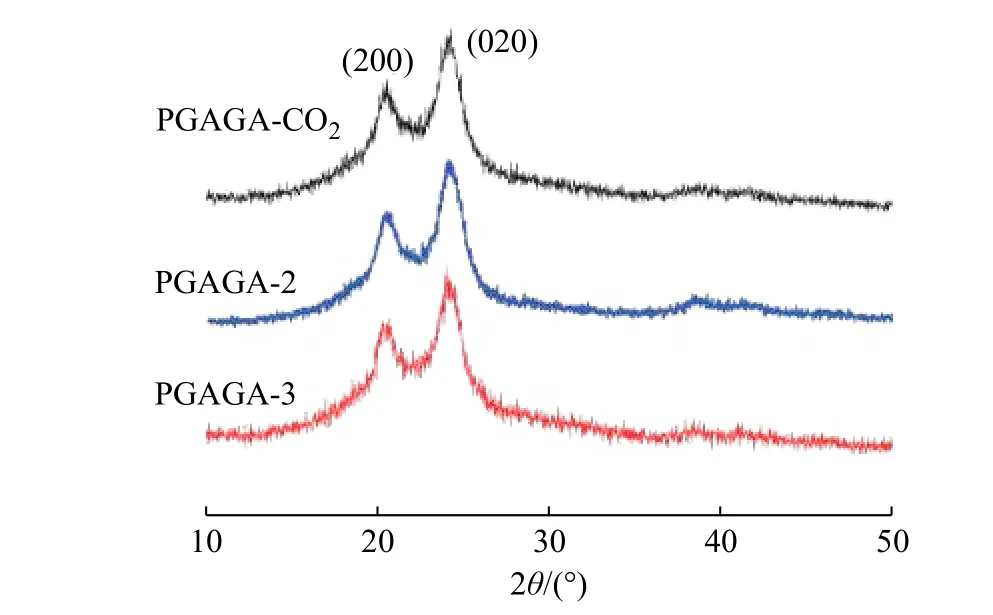
图 3 PGABA 的WAXD 谱图Fig. 3 WAXD patterns of PGABA
PGABA 是由2-吡咯烷酮阴离子开环聚合得到的,其分子量与产率主要取决于催化剂和引发剂的浓度。前期研究表明,与酰基化合物引发剂不同,CO2不直接参与聚合,只对聚合产生促进作用,其机理如图4 所示[19]。在步骤1 中,2-吡咯烷酮(1)与碱反应生成吡咯烷酮盐(2)的溶液后,以其作为聚合反应催化剂。引入CO2后会先与催化剂2 反应生成2-吡咯烷酮的羧酸盐(3)。如图4 中的步骤2 所示,在反应体系中1、2、3 同时存在,而3 形成的阴阳离子对较松散,有利于单体在催化剂2 的作用下连续开环插入,同时CO2还能清除单体分解形成的链终止剂氨基丁酸,从而进一步促进聚合[16]。由上述机理可知,CO2的加入会使体系中游离的催化剂2 浓度降低,进而影响聚合进程。

图 4 CO2 存在下的2-吡咯烷酮聚合的机理[20]Fig. 4 Polymerization mechanism of 2-pyrrolidone in the presence of CO2[20]
需要说明的是,当催化剂摩尔分数为6%时,若无引发剂存在,仅通入CO2无法得到聚合产物。由于CO2与引发剂B 作用机理不同,CO2的加入会与体系中游离的催化剂2 反应生成3,从而使2 的浓度降低,导致第2 步聚合缓慢,聚合24 h 仍没有产物。当催化剂摩尔分数提高为8%时,CO2与2 反应生成羧酸盐3 后,仍有足够的2 来催化聚合反应,聚合24 h后可得到产物PGABA-CO2。
由表1 可知,相同聚合时间下仅通入CO2制备的PGABA-CO2产率为24.4%,而仅使用引发剂得到的PGABA-1 产率为80.0%,表明酰基引发剂有利于提高产率。图5 给出了PGABA-CO2和PGABA-1的GPC 淋洗曲线。与PGABA-CO2相比,PGABA-1的分子量分布更宽。这主要是由于一方面酰基引发剂的作用效率高于CO2[16],单体转化率高,聚合速率较快,同时本体聚合自身热效应又加快了反应速率,导致酰基引发体系的黏度很快达到极限,分子链和单体扩散困难[20];另一方面PGABA 极易结晶,短时间内形成的大量微晶遮蔽活性中心影响链增长。
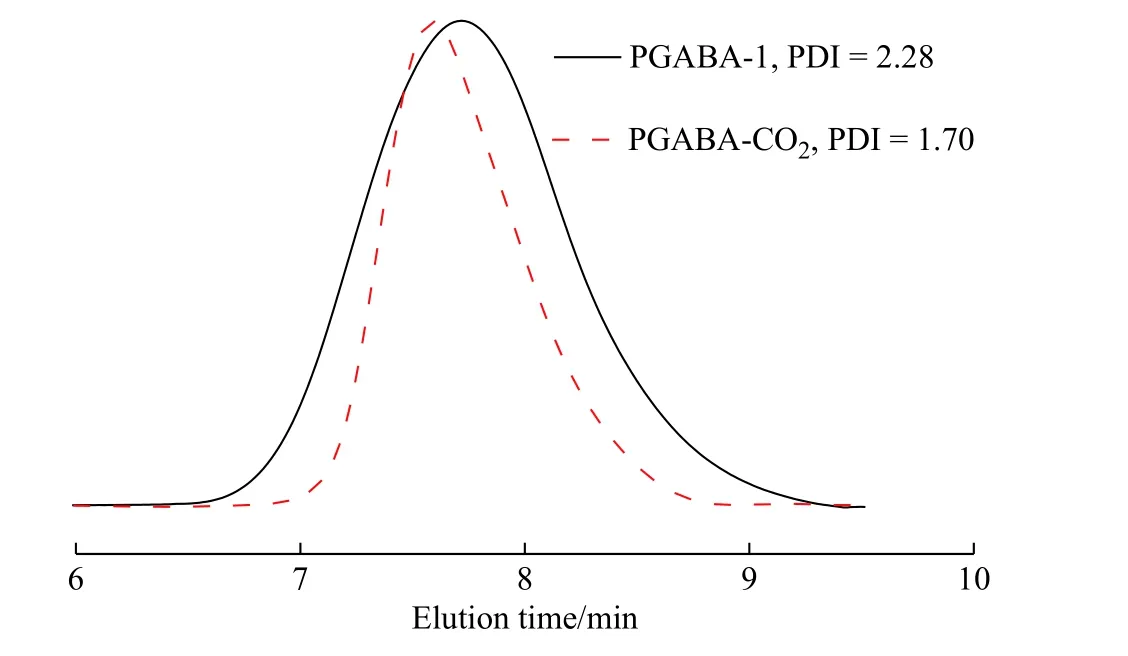
图 5 PGABA 的GPC 淋洗曲线Fig. 5 GPC elution curves of PGABA
此外,由表1 可知,相同聚合时间下,当催化剂的摩尔分数为6%时,引入CO2后产物的黏均分子量从χ(CO2)=0 时的5.0×103升高到χ(CO2)=3.3%时的1.4×104,然后逐渐降低,产率则从80.0%降到CO2用量最大时的31.7%。当催化剂的摩尔分数为7%时,随χ(CO2)的增加,产物的黏均分子量也呈现先升高后降低的趋势,产率也逐步降低。当催化剂摩尔分数提高到9%时,χ(CO2)的增加对产物的黏均分子量和产率的影响作用减弱,黏均分子量变化不大,产率则从54.3%降为48.4%。上述数据说明,在使用适量引发剂的体系内通入适量的CO2可以提高产物的黏均分子量,但同时导致了产率的降低。尽管如此,与仅使用CO2的体系(PGABA-CO2)相比,产率仍有所提高。可见,CO2具有清除反应体系中链终止剂和加快聚合反应速率的作用,因此适量CO2的加入有助于延长活性种寿命,提高产物的黏均分子量。另一方面,CO2的加入会与2 形成2-吡咯烷酮羧酸盐3,从而降低体系中催化剂的含量,因此过量的CO2不利于产物的黏均分子量的提高。由文献[16]可知,CO2的促进效率低于酰基引发剂,随着CO2用量的增加,体系内2-吡咯烷酮羧酸盐3 的比例增加,由其促进生成的活性种比例增加,其活性种链增长速率比酰基引发剂生成的活性种增长速率缓慢,同时催化剂2 的浓度也降低,因此随着CO2用量的增加,相同聚合时间下的产率降低。
比较催化剂用量的影响可见,在引发剂和CO2共同作用下,当χ(CO2)<13.2%时,增加催化剂的用量,产率和黏均分子量均有先增加后降低的趋势。当χ(CO2)=13.2%时,产率提升。尽管此时黏均分子量也呈现先增加后降低的趋势,但黏均分子量提升的幅度远低于CO2用量较小的体系。由前述机理可知,体系中催化剂含量越高或CO2用量越大,3 的含量越高,聚合反应速率越快。由于PGABA 分子链中氢键密度高,特别容易结晶,在CO2用量大和催化剂浓度高的情况下聚合速率过快,快速生成的低分子量PGABA 发生结晶固化,大量链末端活性种被包埋,阻止了单体与生长中心接触,链增长困难,分子量难以提高。
2.3 热性能分析
图6(a)为PGABA 样品的TG 曲线,图6(b)为PGABA 样品的微熵热重(DTG)曲线,结果列于表2。将热分解质量损失5%时的温度(T5)定为初始降解温度,热降解最大失重速率温度记为Tp。由图6(a)可知,仅使用CO2制备的产物PGABA-CO2比仅由引发剂引发聚合的产物PGABA-1 的分解速率低。当引发剂和CO2共同作用、催化剂摩尔分数为9%时,随着CO2用量的增加,产物分解速率降低。PGABA-CO2和PGABA-1 具有相似的分子量,其T5分别为266、263 ℃,Tp分别为306、302 ℃。PGABA-CO2的T5和Tp均比PGABA-1 的相应值高,说明CO2的引入提高了产物的热稳定性。使用CO2促进剂代替引发剂时聚合产物的分子量分布较窄,热稳定性更佳。当催化剂的摩尔分数为9%时制得的PGABA 黏均分子量低于PGABA-CO2和PGABA-1 的。当χ(CO2)<13.2%时,由引发剂B 引发聚合的效应占主导地位,相比未使用CO2的PGABA-1,PGABA-10 和PGABA-9 的热稳定性没有明显提升。随着χ(CO2)增加到13.2%,CO2在聚合中的效应增强,PGABA-11的T5升至279 ℃,与其熔点265 ℃相差14 ℃,Tp为311 ℃,均高于其他样品的相应值,且最大分解速率降低,分解温度区间变宽,热稳定性得到大幅提升,扩大了熔融加工的操作窗口,有利于PGABA 的熔融加工。
由图6(b)可知,PGABA 的DTG 曲线存在2 个峰,表明热分解过程非1 级反应。这是由于PGABA 的热分解过程包括链中无规降解和从链端开始解聚2 种方式,且这2 种分解方式的反应速率不同。无规降解不仅降低聚合物分子量,而且增加链端数目,从而加速链端解聚[21],故在热降解过程中占主导地位,因此这2 个峰分别为PGABA 无规降解和链端解聚的峰。

图 6 PGABA 样品在氮气氛中的TG(a)和DTG(b)曲线Fig. 6 TG (a) and DTG (b) curves of PGABA in nitrogen atmosphere
PGABA 样品的DSC 测试结果见图7,相关数据列于表3 中。图7(a)为PGABA 样品第1 次升温的DSC曲线,由图可知各PGABA 样品的熔点(Tm1)均在265 ℃附近,改变催化剂和CO2用量对熔点无明显影响。
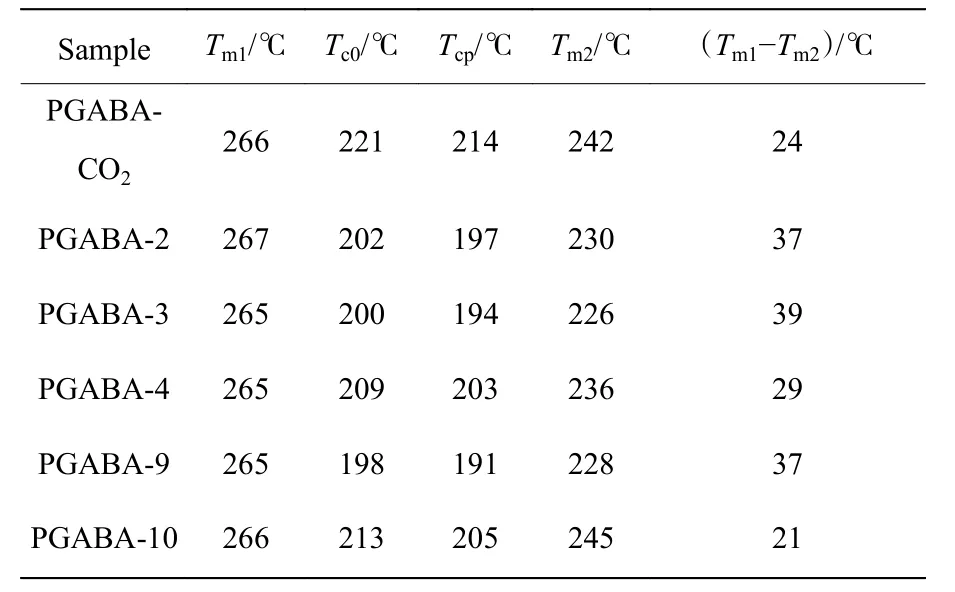
表 3 PGABA 的热力学参数Table 3 Thermodynamic parameters of PGABA

表 2 PGABA 在氮气氛中的热降解温度Table 2 Thermal degradation temperature of PGABA in nitrogen atmosphere
图7(b)为PGABA 样品第1 次降温的DSC 曲线。由TG 测试可知,PGABA 在260 ℃左右开始热分解,因此降温过程中的结晶行为仅作为一种参考。PGABA-CO2的初始结晶温度(Tc0)和结晶峰值温度(Tcp)分别为221 ℃和214 ℃,而PGABA-2 的Tc0和Tcp分别为202 ℃和197 ℃。这说明CO2的引入提高了产物的结晶温度。当催化剂的摩尔分数为6%时,随着χ(CO2)从0 (PGABA-2)增加到6.6% (PGABA-4),Tc0和Tcp分别增加了7 ℃和5 ℃。当催化剂的摩尔分数为9%时,随着χ(CO2)增加,样品PGABA-10 的 Tc0和Tcp比PGABA-9 的Tc0和Tcp分别增加了15 ℃和14 ℃。这说明CO2的存在一定程度上促进了PGABA 的结晶,且随催化剂摩尔分数的增加,促进结晶的趋势增强。
图7(c)为PGABA 样品第2 次升温的DSC 曲线,由于样品在第1 次升温过程中已发生热分解,第2 次升温过程中温度高于260 ℃后的曲线无法得到。与第1 次升温的熔点Tm1相比,第2 次升温时的熔点Tm2均有下降。这是因为断链后PGABA 结晶度和晶体完善程度减弱,使熔点降低。PGABA-CO2和 PGABA-2 相比,Tm2与Tm1的差值分别为24 ℃和37 ℃, PGABA-CO2的Tm2下降幅度较小,说明CO2的引入有助于抑制热分解行为。当催化剂的摩尔分数为6%时,随着CO2用量的增加,PGABA 样品Tm2与Tm1的差值由39 ℃缩小到29 ℃,下降幅度降低。当催化剂摩尔分数为9%时也有同样的趋势,进一步证明了一定范围内的CO2加入有利于提高PGABA 的热稳定性。
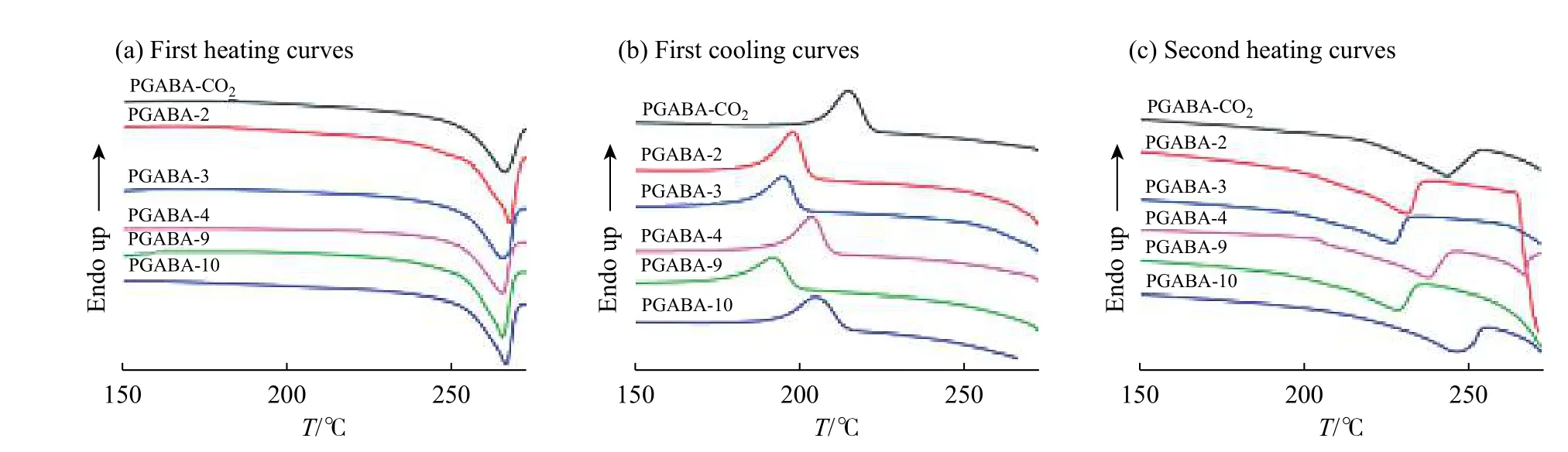
图 7 PGABA 样品的DSC 曲线Fig. 7 DSC curves of PGABA samples
3 结 论
(1)阴离子开环聚合制备PGABA 过程中,在酰基化合物引发聚合的同时引入适量CO2有利于提高产物的分子量,同时引发剂的存在有利于提升CO2聚合体系产物收率。
(2)在引发剂和CO2共同作用下,适当增加催化剂用量可提升产物黏均分子量,而对产率的影响较复杂。
(3)所合成的PGABA 均为α 晶型,改变催化剂、引发剂和CO2的用量对晶型没有影响。
(4)CO2的引入对PGABA 的熔点没有影响,但是会促进结晶,一定程度上使结晶温度升高。
(5)随着CO2用量的增加,PGABA 的热稳定性提升,有利于PGABA 的熔融加工。
猜你喜欢
纺织科学研究(2021年7期)2021-08-14
合成树脂及塑料(2021年6期)2021-01-09
食品与生物技术学报(2020年11期)2020-01-05
中国洗涤用品工业(2017年2期)2017-04-16
现代检验医学杂志(2016年1期)2016-11-12
国外医药(抗生素分册)(2016年4期)2016-07-12
中国洗涤用品工业(2016年2期)2016-02-28
烟草科技(2015年8期)2015-12-20
中国生化药物杂志(2015年4期)2015-07-07
中国医药科学(2014年22期)2014-12-22
