
小麦Pinb基因启动子区CRISPR/Cas9基因编辑载体的构建
2020-07-04 03:04张淑娟张荣志宋国琦李玉莲高洁李玮高世庆李根英
山东农业科学 2020年1期
关键词:小麦
张淑娟 张荣志 宋国琦 李玉莲 高洁 李玮 高世庆 李根英

摘要:CRISPR/Cas9介导的基因组编辑技术已经逐渐成熟并在多种植物中得到成功应用,促进了基因功能的研究。小麦籽粒硬度是决定小麦加工品质的重要性状,Pinb是控制籽粒硬度的关键基因之一。本研究以小麦Pinb基因为对象,联合采用常规PCR和Golden Gate cloning技术,构建了包含小麦Pinb基因启动子区双靶点的CRISPR/Cas9基因编辑载体。将重组载体转入小麦原生质体中进行sgRNA活性的检测,在两个靶点位置均发生了突变,编辑效率分别为4.57%和4.37%,基因编辑类型包括碱基替换和碱基缺失。该研究为在小麦中实现Pinb基因的定点突变奠定了基础,对其功能研究和小麦品质改良均具有重要意义。
关键词:小麦;CRISPR/Cas9;基因编辑;Pinb基因
中图分类号:S512.103.53:Q782 文献标识号:A 文章编号:1001-4942(2020)01-0001-09
Abstract The genome editing technology system mediated by CRISPR/Cas9 has ripen and successfully applied in many plants, which promotes the study of gene function. Hardness of wheat grain is an important trait for determining the quality of wheat processing. The Pinb gene is one of the key genes controlling grain hardness. In the study, we chose the Pinb gene of wheat as research object, and constructed a CRISPR/Cas9 genome editing vector containing dual targets in the Pinb gene promoter region of wheat by conventional PCR and Golden Gate cloning technology. Then we transferred the recombinant vector into wheat protoplast and detected the activity of sgRNA. We found the dual targets position had mutated, and the gene editing types included base substitution and base deletion, and the corresponding editing efficiencies were 4.57% and 4.37%, respectively. The results laid the foundation for realizing the site-directed mutagenesis of the Pinb gene, which was of great significance for the functional study of Pinb gene and the improvement of wheat quality.
Keywords Wheat; CRISPR/Cas9; Gene editing; Pinb gene
CRISPR/Cas9 (clustered regularly interspaced short palindromic repeats/CRISPR-associated nuclease 9)作为一种新兴的基因编辑系统,具有载体构建相对简单、靶向特异性高、可以多处打靶等特点,在基因功能研究方面逐渐成熟并成功应用于多种动植物。其原理是crRNA与tracrRNA通过碱基配对结合形成crRNA/tracrRNA复合体(可以人工设计改造这两种RNA,形成单个具有引导作用的sgRNA),引导Cas9蛋白在与crRNA配对靶位点处形成双链断裂(double-stranded DNA breaks,DSBs),之后细胞可以通过非同源性末端接合(non-homologous end jouning,NHEJ)修复机制实现基因组特定位点的DNA插入、缺失、碱基突变或修饰,从而使基因发生移码突变。另外,还可以通过同源重组等方式进行精准基因编辑。
目前植物CRISPR/Cas9系统日趋完善,已经在多个物种中成功实现了定点基因组编辑,除在拟南芥[1,2]、烟草[1,3]等模式植物上获得成功,在水稻[4-8]、大豆[9-11]、高粱[1]、玉米[12-15]、番茄[16]、大麥[17]、甜橙[18]、西瓜[19]、葡萄[20]、小麦[21-24]等农作物上也成功实现了CRISPR/Cas9的应用。普通小麦 (Triticum aestivum L.) 是世界上种植面积最广的作物之一,具有非常复杂的多倍体基因组(17 Gb)和高比例的重复序列(>80%),为小麦遗传和功能分析带来了挑战。目前,在普通小麦中只有少数使用CRISPR/Cas9进行基因编辑的报道。由CRISPR/Cas9系统获得的TaMLO突变体具有对白粉病的广谱抗性[22]。 Zhang等[25]将CRISPR/Cas9编辑方法用于六倍体面包小麦和四倍体硬粒小麦,产生的突变体没有任何可检出的转基因。Zhang等[26]利用CRISPR/Cas9系统同时敲除小麦EDR1基因的三个同源基因,获得的植株对白粉病具有很好的抗性。另外,利用CRISPR/Cas9技术可以有效地减少小麦籽粒中α-醇溶蛋白含量,从而为谷蛋白不耐受的消费者提供特殊面包和硬质小麦系列[23]。
籽粒硬度(grain hardness)是重要的小麦品质性状之一,对谷物加工品质具有重要影响。小麦籽粒硬度主要由位于5D染色体短臂上的2个主效基因Pina(Puroindoline a)和Pinb(Puroindoline b)调控,并受Gsp-1基因(Grain Softness Protein)的影响。Pina和Pinb紧密连锁,编码区均为447 bp,没有内含子,共同形成小麦籽粒硬度的分子遗传基础。Pina和Pinb基因的突变或缺失均会引起小麦胚乳质地的硬度增加。尽管二倍体小麦祖先种A和B中存在Pina和Pinb基因,但是四倍体驯化小麦中却不存在这两个基因,所以表现出硬粒表型。而六倍体小麦由于D组的加入,重新获得了Pina和Pinb,故而表现出籽粒硬度的多样性。所以小麦的硬度基因位点从一定程度上反映了小麦的进化和驯化过程。
目前,关于小麦硬度基因表达调控的研究较少。小麦Pina和Pinb基因的启动子已经分离出来,且有关于启动子组成的分析,但是在启动子调控方面并没有相关数据。使用CRISPR/Cas9基因编辑技术编辑农作物相关基因的启动子,可对作物数量性状产生微妙的影响。Rodriguez-Leal等[27]利用CRISPR/Cas9体系改良番茄数量性状,通过定点修饰顺式调控序列实现对数量性状的精准调控。挖掘和创制顺式调控序列的变异,对于作物遗传改良意义重大。
利用CRISPR/Cas9突变相关基因的启动子而不是这些基因本身,能够实现对数量性状的精细调节,微调基因表达而不是剔除或灭活它们编码的蛋白。本研究拟对TaPinb基因的启动子区域进行基因编辑,目的在于进一步明确TaPinb基因的功能和其在籽粒质地形成中的作用,并为通过基因工程方法快速改良我国现有小麦优良品种的籽粒硬度提供有用信息。
1 材料与方法
1.1 试验材料
植物CRISPR/Cas9基因编辑载体为pYLCRISPR/Cas9PubiB,GenBank登录号为KR029110.1,它是在双元载体pCAMBIA1300(AF234296)的基础上改造而来的,Cas9p模拟了禾本科植物基因具有5′端GC含量较高的特征,是设计合成的植物优化密码子基因。sgRNA的载体pYPQ131D-TaU6和pYPQ132C-TaU6是CRISPR/Cas9基因编辑载体的中间载体,均含有小麦U6启动子TaU6,用以启动sgRNA。以上质粒均由本实验室保存。
T4连接酶、DNA Marker DL2000等试剂购自TaKaRa公司;FastPfu、2×Taq Mix、DH5α等购自北京全式金公司;限制性内切酶BgⅢ、BsmBⅠ等购自Fermentas公司;BsaⅠ-HF购自NEB公司;引物由青岛擎科梓熙生物技术有限公司合成。
1.2 表达载体的构建
1.2.1 Pinb基因的扩增和sgRNA的选择 以Pinb-F和Pinb-R为引物,以小麦品种Fielder的基因组DNA为模板,用高保真酶扩增Pinb基因,PCR体系为:5×buffer 10 μL,dNTP(2.5 mmol/L)5 μL,FastPfu 1 μL,上下游引物(10 μmol/L)各2 μL,加水补齐至50 μL。程序为:95℃ 2 min;95℃ 20 s,56℃ 50 s,72℃ 30 s,33个循环;72℃ 5 min。电泳切胶回收,连接载体,送测序公司测序。
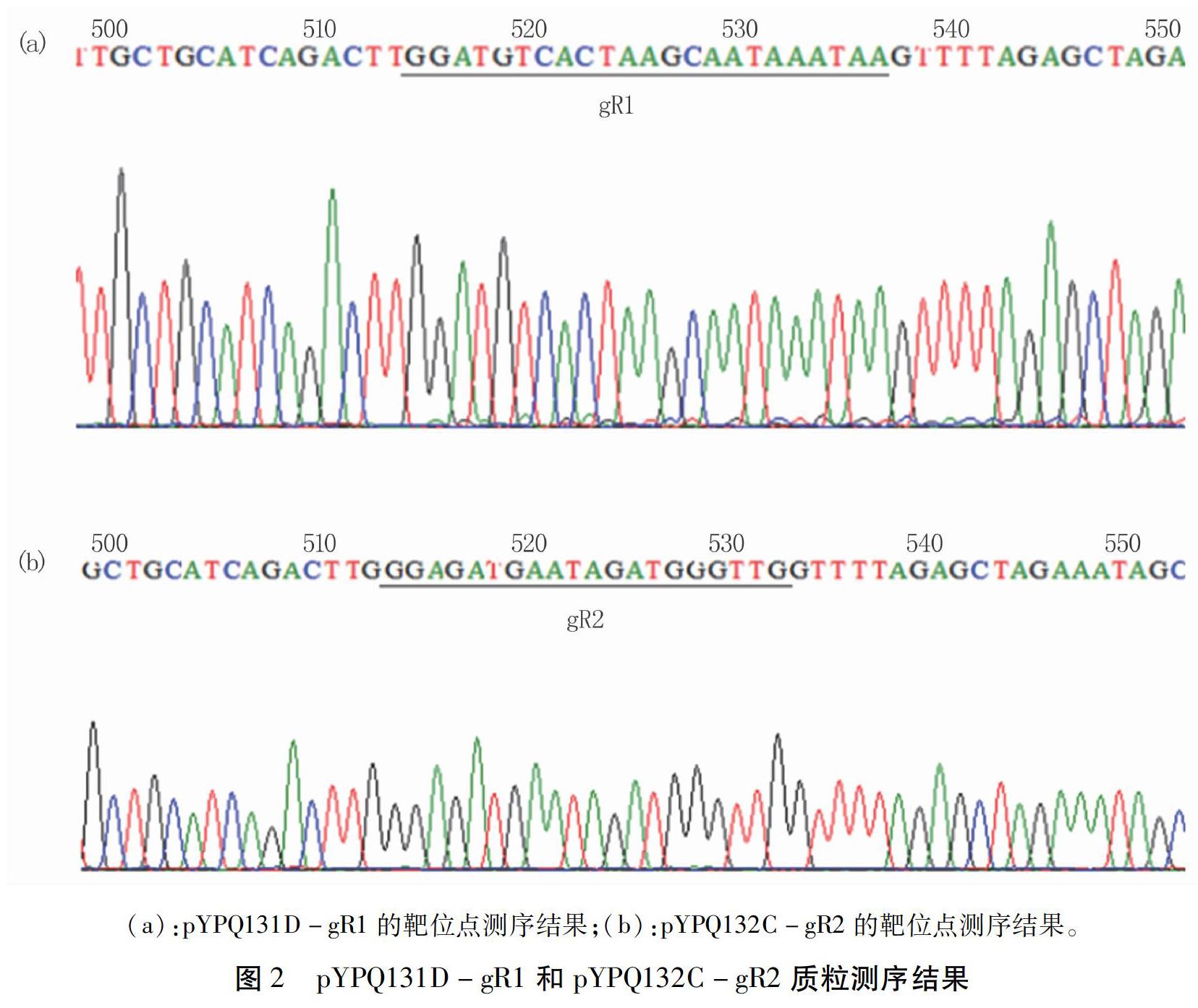
通过网站CRISPRdirect (http://crispr.dbcls.jp/)在TaPinb基因ATG前的啟动子区域寻找2个适合的打靶位点,在PAM结构前选择合适的序列片段设定为靶序列,分别为:sgRNA1:5′-ATGTCACTAAGCAATAAATAA-3′;sgRNA2:5′-GGAGATGAATAGATGGGTTG-3′。
1.2.2 sgRNA表达盒构建方法
(1)线性化克隆载体pYPQ131D、pYPQ132C:使用限制性内切酶BgⅢ和BsmBⅠ双酶切克隆载体pYPQ131D-TaU6、pYPQ132C-TaU6,37℃温育1 h,电泳后利用DNA回收试剂盒回收3.5 kb的线性化片段,定量后备用。
(2)引物磷酸化退火形成双链:Pinb基因的sgRNA1的引物对应的是Pinb-TaU6-gR1F和Pinb-TaU6-gR1R(表1),sgRNA2的引物对应的是Pinb-TaU6-gR2F和Pinb-TaU6-gR2R(表1),引物中的下划线碱基部分分别与pYPQ131D和pYPQ132C载体中的序列匹配。分别将sgRNA1和sgRNA2的2条引物进行磷酸化,直接退火形成双链,程序在PCR仪中按下列条件运行:37℃ 30 min,95℃ 5 min,5℃/min (0.08℃/s)的速度将温度降至25℃。或者是沸水中煮5 min,之后自然冷却至室温。
(3) 连接转化:将上述线性化的克隆载体pYPQ131D、pYPQ132C,和磷酸化后的寡核苷酸链sgRNA1和sgRNA2分别在16℃过夜连接,转化DH5α,平板四环素抗性。之后挑平板单克隆送测序,验证是否插入,最终得到pYPQ131D-gR1和pYPQ132C-gR2。
1.2.3 组装sgRNA表达盒到pYLCRISPR/Cas9载体
(1)sgRNA表达盒的扩增:以上述试验中获得的pYPQ131D-gR1和pYPQ132C-gR2为模板,分别以YL-131D-F和YL-131D-R、YL-132C-F和YL-132C-R为引物,用高保真酶扩增TaU6-gR1和TaU6-gR2这2个表达盒,PCR体系为:5×buffer 10 μL,dNTP(2.5 mmol/L)5 μL,FastPfu 1 μL,上下游引物(10 μmol/L)各2 μL,加水补齐至50 μL。程序为:95℃ 2 min; 95℃ 20 s,62℃ 20 s,72℃ 30 s,33个循环;72℃ 5 min。电泳切胶回收,定量备用。
(2) pYLCRISPR/Cas9载体与sgRNA表达盒的酶切连接反应:利用Golden Gate cloning的方法,将pYPQ131D-sgRNA1和pYPQ132C-sgRNA2这两个表达盒克隆到pYLCRISPR/Cas9载体中。连接用的2个表达盒即上述步骤中的TaU6-gR1和TaU6-gR2。酶切连接反应体系为:10×Cutsmart Buffer 1 μL ,10 mmol/L ATP 1 μL ,TaU6-gR1 1 μL,TaU6-gR2 1 μL,pYLCRISPR/Cas9Pubi-B 2 μL,BsaⅠ-HF 1 μL,T4 ligase 1 μL,ddH2O补足10 μL。用变温循环进行酶切连接约10~15循环: 37℃ 5 min, 10℃ 5 min, 20℃ 5 min;最后37℃ 5 min。
(3)连接转化:将连接产物转化大肠杆菌,重组质粒经转化到DH5α感受态细胞,之后于LB(Kan)平板上培养至克隆长出后,挑单克隆进行菌液PCR鉴定及测序。引物用的是SP-L1和SP-R。
1.3 小麦原生质体的提取和转化
参照文献[28]的小麦原生质体制备及转化方法并适当调整。种子在25℃(16L/8D)培养条件下生长7~10 d。取15~20片新鲜叶片用手术刀片切成0.5~1.0 mm宽的细条并转移到10 mL酶解液(1.5%纤维素酶R10,0.75%离析酶,0.6 mol/L甘露醇,20 mmol/L MES,10 mmol/L KCl,10 mmol/L CaCl2,0.1% BSA)。将酶解液和叶条置于50 r/min摇床上,用锡箔纸包裹在黑暗条件下酶解6 h。酶解后,加入等体积W5(2 mmol/L MES, 154 mmol/L NaCl, 125 mmol/L CaCl2, 5 mmol/L KCl) 轻柔摇晃释放出原生质体,清洗3次。用70 μm的细胞网筛过滤酶解液,用50 mL圆底离心管收集滤液。加入MMG溶液重悬(0.4 mol/L mannitol, 15 mmol/L MgCl2, 4 mmol/L MES)。将构建好的质粒利用PEG介导的方法转化入原生质体。最后,黑暗或者弱光下25℃培养原生质体2~3 d。离心后收集小麦原生质体,用DNA快速提取试剂盒提取基因组DNA。
1.4 基因编辑结果检测
使用特异性引物对靶位点区域进行扩增。将正向和反向的Barcode分别添加到PCR引物的5′端用于文库构建(引物为表1中的PF和PR,下划线为Barcode)。通过具有相应Barcode的引物扩增对应于Pinb基因2个靶位点的区域。扩增后,将等量的PCR产物混合,按照German等[29]的方法构建2×150 bp paired-end 文库,用于诺禾致源公司进行Illumina测序。在靶基因的靶向位点处发生的InDel(包括替换、插入和缺失)被认为是突变。
2 结果与分析
2.1 靶向TaPinb的sgRNA设计
从小麦品种Fielder里扩增TaPinb基因的序列,扩增序列见图1,与NCBI数据库中该基因序蓝色代表TaPinb基因的编码区,下划线标记序列为靶向TaPinb基因的两个sgRNA序列。
图1 TaPinb基因及sgRNA序列列100%相似。TaPinb基因全长447个碱基,没有内含子,应用软件CRISPRdirect在TaPinb基因ATG前的启动子区域找到两个合适的sgRNA,分别为:sgRNA1:5′-ATGTCACTAAGCAATAAATAA-3′;sgRNA2:5′-GGAGATGAATAGATGGGTTG-3′。
2.2 sgRNA连接到pYPQ31D和pYPQ132C
分别将sgRNA1和sgRNA2的2条引物进行磷酸化,直接退火形成双链,然后分别与用限制性内切酶BgⅢ和BsmBⅠ双酶切后的克隆载体pYPQ131D-TaU6、pYPQ132C-TaU6连接转化,测序结果见图2。
2.3 TaPinb的sgRNA表达盒的扩增
以pYPQ131D-gR1和pYPQ132C-gR2为模板,分别以YL-131D-F和YL-131D-R、YL-132C-F和YL-132C-R为引物,用高保真酶扩增TaU6-gR1和TaU6-gR2这2个表达盒,扩增产物大小为571 bp(图3)。
2.4 sgRNA表达盒和Cas9双元表达载体的组合
我们应用Golden Gate cloning技术,一步完成sgRNA表达盒的拼接及与Cas9双元表达载体的组合,从而获得CRISPR/Cas9编辑载体(图4)。如图5所示,用SP-L1和SP-R进行PCR扩增,可见约1.5 kb的条带,包括2个sgRNA表达盒的大小(TaU6-sgRNA),与预期相符,而对照pYLCRISPR/Cas9Pubi-B双元表达载体扩增出的条带是ccdB,约657 bp,这说明sgRNA1和sgRNA2表達盒已经组装进入pYLCRISPR/Cas9Pubi-B双元表达载体,获得了TaPinb的CRISPR/Cas9基因编辑载体。
为了进一步验证TaPinb的CRISPR/Cas9基因编辑载体是否构建成功,我们将获得的质粒送测序公司进行测序,引物用的是SP-L1和SP-R,结果检测到了2个sgRNA的靶序列,同时在靶序列上游都检测到了TaU6启动子序列,说明sgRNA1和sgRNA2表达盒构建成功,并成功地组装进入pYLCRISPR/Cas9Pubi-B双元表达载体,证明了Pinb的CRISPR/Cas9基因编辑载体构建成功(图6)。
采用PEG法转化小麦原生质体,将转化后孵育2~3 d的小麦原生质体用植物基因组DNA提取试剂盒提取小麦基因组DNA,对靶位点进行扩增后建库测序。测序结果表明目标的编辑类型主要是SNP和InDel。其中,Pinb-gR1位点具有4.57%的突变效率,包括4.49%的SNP和0.08%的InDel。Pinb-gR2位点的突变效率4.37%,包括4.05%的SNP和0.33%的InDel(表2)。Pinb-gR1突变位点检测(图7a)发现编辑位点(PAM序列前第4位)处有碱基的替换和缺失,包括单碱基的替换、单碱基的缺失以及4个碱基的缺失。Pinb-gR2突变位点检测结果见图7b,突变类型主要是单碱基替换和缺失,另外,在PAM前的其它位点也检测到了突变。
3 討论与结论
普通小麦是世界上种植面积最广的作物之一,籽粒硬度是决定小麦加工品质最重要的性状之一,对小麦出粉率、淀粉损伤和生面粉团的吸水性都具有较大影响。因此Pin基因的功能研究和转基因应用对小麦品质改良理论与实践具有重要意义,为进一步研究小麦中Pin基因在影响籽粒硬度的分子机理奠定基础,为选育优质专用小麦提供种质资源。
基因敲除是研究基因功能最简单的方法之一。CRISPR/Cas9系统能够诱导基因组产生双链断裂,通过细胞自身的NHEJ修复途径造成若干碱基的插入、缺失和替换,从而造成基因的移码突变,起到基因敲除的作用。目前植物CRISPR/Cas9系统已日趋完善,已经在多种植物中成功实现了定点基因组编辑。
为了探索CRISPR/Cas9系统应用于小麦基因组编辑的效率和特异性,本研究针对Pinb基因的启动子区设计了两个sgRNA。本载体系统可采用Golden Gate cloning技术[30], 其原理是利用BsaⅠ(type Ⅱs restriction enzyme)的识别位点和切割位点不重叠的特性,可以设计出多种不同的且非回文结构的粘性末端。多个要连接的片段在连接点具有特异的互补末端可以连接,而非连接点的末端之间不互补不能连接(相同末端分子间也不互补不能连接),因此连接反应是向着目标单方向进行,效率很高。
我们通过PEG介导的原生质体转化方法将构建好的CRISPR/Cas9植物表达载体转入小麦的原生质体,Illumina测序结果显示CRISPR/Cas9系统在小麦原生质体中进行了Pinb基因编辑。在两个靶位点均检测到了碱基的突变,编辑效率分别为4.57%和4.37%,其中多数情况是碱基替换,少数情况是碱基的删除。另外,在小麦原生质体编辑情况检测中发现除了PAM序列前第四个碱基处发生突变外,在gRNA靶标区域的其它碱基处也发现了一些突变,这可能是由于原生质体DNA制备过程有损伤,导致测序出现个别碱基的差异。有研究表明CRISPR/Cas9的特异性仅与gRNA配对的靠近PAM处7~12 bp碱基相关,其它碱基的突变是否与Cas9的特异性有关也需要在转基因小麦中进一步验证。
本研究通过PEG介导的方法将构建好的CRISPR/Cas9基因编辑载体转入小麦原生质体,利用高通量测序检测到了靶标基因的编辑,证明该载体可以应用于小麦的基因组编辑,从而为应用CRISPR/Cas9系统获得小麦基因组编辑植株打下坚实的基础。目前我们正在将这个载体进行农杆菌介导的小麦遗传转化,下一步将会在转基因小麦植株中检测编辑类型。
参 考 文 献:
[1] Jiang W, Zhou H, Bi H, et al. Demonstration of CRISPR/Cas9/sgRNA-mediated targeted gene modification in Arabidopsis, tobacco, sorghum and rice [J]. Nucleic Acids Res., 2013, 41(20):e188.
[2] Li J F, Norville J E, Aach J, et al. Multiplex and homologous recombination-mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9 [J]. Nat. Biotechnol., 2013, 31(8):688-691.
[3] Nekrasov V, Staskawicz B, Weigel D, et al. Targeted mutagenesis in the model plant Nicotiana benthamiana using Cas9 RNA-guided endonuclease [J]. Nat. Biotechnol., 2013, 31(8):691-693.
[4] Zhang H, Zhang J, Wei P, et al. The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation [J]. Plant Biotechnol. J., 2014, 12(6):797-807.
[5] Zhou H, Liu B, Weeks D P, et al. Large chromosomal deletions and heritable small genetic changes induced by CRISPR/Cas9 in rice [J]. Nucleic Acids Res., 2014, 42(17):10903-10914.
[6] Ma X, Zhang Q, Zhu Q, et al. A robust CRISPR/Cas9 system for convenient, high-efficiency multiplex genome editing in monocot and dicot plants [J]. Mol. Plant, 2015, 8(8):1274-1284.
[7] Xu K, Ren C, Liu Z, et al. Efficient genome engineering in eukaryotes using Cas9 from Streptococcus thermophilus[J]. Cell Mol. Life Sci., 2015, 72(2):383-399.
[8] Li J, Sun Y, Du J, et al. Generation of targeted point mutations in rice by a modified CRISPR/Cas9 system [J]. Mol. Plant, 2017, 10(3):526-529.
[9] Jacobs T B, LaFayette P R, Schmitz R J, et al. Targeted genome modifications in soybean with CRISPR/Cas9 [J]. BMC Biotechnol., 2015, 15:16.
[10] Sun X, Hu Z, Chen R, et al. Targeted mutagenesis in soybean using the CRISPR-Cas9 system [J]. Sci. Rep., 2015, 5:10342.
[11] Tang F, Yang S, Liu J, et al. Rj4, a gene controlling nodulation specificity in soybeans, encodes a thaumatin-like protein but not the one previously reported [J]. Plant Physiol., 2016, 170(1):26-32.
[12] Liang Z, Zhang K, Chen K, et al. Targeted mutagenesis in Zea mays using TALENs and the CRISPR/Cas system [J]. J. Genet. Genomics, 2014, 41(2):63-68.
[13] Xing H L, Dong L, Wang Z P, et al. A CRISPR/Cas9 toolkit for multiplex genome editing in plants [J]. BMC Plant Biol., 2014, 14:327.
[14] Svitashev S, Young J K, Schwartz C, et al. Targeted mutagenesis, precise gene editing, and site-specific gene insertion in maize using Cas9 and guide RNA [J]. Plant Physiol., 2015, 169(2):931-945.
[15] Char S N, Neelakandan A K, Nahampun H, et al. An Agrobacterium-delivered CRISPR/Cas9 system for high-frequency targeted mutagenesis in maize [J]. Plant Biotechnol. J., 2017, 15(2):257-268.
[16] Brooks C, Nekrasov V, Lippman Z B, et al. Efficient gene editing in tomato in the first generation using the clustered regularly interspaced short palindromic repeats/CRISPR-associated9 system [J]. Plant Physiol., 2014, 166(3):1292-1297.
[17] Lawrenson T, Shorinola O, Stacey N, et al. Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA-guided Cas9 nuclease [J]. Genome Biol., 2015, 16:258.
[18] Jia H, Wang N. Targeted genome editing of sweet orange using Cas9/sgRNA [J]. PLoS ONE, 2014, 9(4):e93806.
[19] Tian S, Jiang L, Gao Q, et al. Efficient CRISPR/Cas9-based gene knockout in watermelon [J]. Plant Cell Rep., 2017, 36(3):399-406.
[20] Wang X, Tu M, Wang D, et al. CRISPR/Cas9-mediated efficient targeted mutagenesis in grape in the first generation [J]. Plant Biotechnol. J., 2017.
[21] Shan Q, Wang Y, Li J, et al. Targeted genome modification of crop plants using a CRISPR-Cas system [J]. Nat. Biotechnol., 2013, 31(8):686-688.
[22] Wang Y, Cheng X, Shan Q, et al. Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew [J]. Nat. Biotechnol., 2014, 32(9):947-951.
[23] Snchez-León S, Gil-Humanes J, Ozuna C V, et al. Low-gluten, nontransgenic wheat engineered with CRISPR/Cas9 [J]. Plant Biotechnol. J., 2018, 16(4):902-910.
[24] Kim D, Alptekin B, Budak H. CRISPR/Cas9 genome editing in wheat [J]. Funct. Integr. Genomics, 2018, 18(1):31-41.
[25] Zhang Y, Liang Z, Zong Y, et al. Efficient and transgene-free genome editing in wheat through transient expression of CRISPR/Cas9 DNA or RNA [J]. Nat. Commun., 2016, 7:12617.
[26] Zhang Y, Bai Y, Wu G, et al. Simultaneous modification of three homoeologs of TaEDR1 by genome editing enhances powdery mildew resistance in wheat [J]. Plant J., 2017, 91(4):714-724.
[27] Rodriguez-Leal D, Lemmon Z H, Man J, et al. Engineering quantitative trait variation for crop improvement by genome editing [J]. Cell, 2017, 171(2):470-480.
[28] Shan Q, Wang Y, Li J, et al. Genome editing in rice and wheat using the CRISPR/Cas system [J]. Nat. Protoc., 2014, 9(10):2395-2410.
[29] German M A, Luo S, Schroth G, et al. Construction of Parallel Analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome [J]. Nat. Protoc., 2009, 4(3):356-362.
[30] Engler C, Kandzia R, Marillonnet S. A one pot, one step, precision cloning method with high throughput capability [J]. PLoS ONE, 2008, 3(11):e3647.
猜你喜欢
飞天(2020年9期)2020-09-06
农民致富之友(2020年8期)2020-05-11
启蒙(2020年2期)2020-02-12
农民致富之友(2019年32期)2019-11-23
清明(2019年6期)2019-11-21
新农村(2017年17期)2017-08-23
农产品市场周刊(2014年22期)2014-08-22
小小说月刊·下半月(2014年8期)2014-05-14
农民科技培训(2014年4期)2014-04-23
少年文艺·我爱写作文(2009年5期)2009-06-08
