
C4ST-1 在大肠杆菌中的可溶性表达
2020-09-10 06:23周正雄堵国成李江华
食品与生物技术学报 2020年6期
李 青, 周正雄, 堵国成,2, 李江华*,2, 康 振,2
(1. 江南大学 生物工程学院,江苏 无锡214122;2. 江南大学 糖化学与生物技术教育部重点实验室,江苏 无锡214122)
硫酸软骨素(chondroitin sulfate,CS)是一类由葡萄糖醛酸和GalNAc 交替组成,并在GalNAc 不同位点硫酸化的线性阴离子多糖[1]。 CS 为防治关节病[2-3]和修复受损的中枢神经系统[4-5]的最佳药物,同时广泛地应用于食品领域[6]。 CS 根据其硫酸化位点不同可分为CSA、硫酸软骨素C(chondroitin sulfate C,CSC)等[7-8]。 目前,CSA 主要从气管、鲨鱼软骨[9]等动物软骨组织中提取得到。 但由于动物组织中提取的硫酸软骨素存在过硫酸化、 产品难以保证一致性、可能携带潜在致病因子等因素[10]限制了CSA 的进一步应用。 作者在前期研究中提出两步法得到CSA。首先,以Bacillus subtilis 为宿主发酵生产软骨素[11];然后以Pichia pastoris 发酵生产的C4ST-1 催化软骨素形成结构单一的CSA[12]。
但相比于Pichia pastoris,E. coli 具有生长速度快,培养简单,遗传背景清楚等优点,其成为重组蛋白表达的首选。 但该系统缺乏蛋白质翻译后修饰系统,表达的C4ST-1 经常错误折叠成形成包涵体[12-13]。在本课题组前期工作中, 将来源于小鼠的C4ST-1在E. coli 中成功地进行了表达,表达产物基本上都以包涵体形式存在。 C4ST-1 含3 对二硫键, 在E.coli 胞质还原性环境中可能会使C4ST-1 的二硫键无法折叠或不正确折叠。 近年来,人们利用硫氧还蛋白还原酶(TrxB)和谷胱甘肽还原酶(Gor)双突变工程菌株[14-17],融合氧化性硫氧还蛋白A(TrxA)[18],或者引进二硫键从头形成体系[19]等策略,在E. coli胞质中成功实现了多种二硫键蛋白的表达。 有文献报道表明Orgami(DE3)等工程菌株不能催化二硫键的从头合成,蛋白质产率可能较低[20]。 Erv1p(EC 1.8.3.2)可以催化二硫键从头形成,向E. coli 细胞质引入二硫键从头形成体系,可能会大幅提高活性蛋白质的产率[20-24]。 DsbC(EC 5.3.4.1)在细胞质过量表达能够促进外源蛋白质的二硫键正确配对和异构化,进而提高胞质可溶性蛋白质表达水平[25-26]。 本研究通过在E. coli 中共表达Erv1p、 DsbC 及C4ST-1,以期实现胞内可溶性蛋白质产量和酶活的提高。
1 材料与方法
1.1 材料
1.1.1 宿主和质粒菌株E. coli JM109,E. coli BL21(DE3),Saccharomyces cerevisiae S288C 及质粒pET-32a(+),pRSFDuet1 均为本实验室保存。
1.1.2 主要试剂和仪器质粒DNA 小量制备试剂盒、琼脂糖凝胶DNA 回收试剂盒、酵母基因组提取试剂盒和细菌基因组提取试剂盒,均购自上海生工生物工程股份有限公司;DNA 标准相对分子质量片段、各种限制性内切酶、DNA 连接酶,均购于大连宝生物工程有限公司;一步法克隆试剂盒,购自NEB公司;SDS-PAGE 预制胶, 标准相对分子质量蛋白质,购自Thermo 公司;胰蛋白胨、酵母提取物,购自英国OXOID 公司;对硝基硫酸苯酯(PNPS)、3′-磷酸腺苷-5′-磷酸(PAP),均购自Sigma-Aldrich 公司;软骨素制备参照文献[11],芳基硫磺基转移酶(ASTIV,EC 2.8.2.1)制备参照文献[12],其他试剂均为国产分析纯。
DYY-6D 型琼脂糖水平电泳槽,购自北京六一生物科技有限公司;蛋白胶电泳槽,购自Thermo 公司;VCX750 型超声破碎仪, 购自美国SONICS 公司;UV2450 型紫外可见分光光度计,购自日本岛津公司;Avanti J-26XP 低温高速冷冻离心机, 购自美国Beckman Coulter 公司。
1.1.3 培养基及培养条件LB 液体培养基(g/L):胰蛋白胨10, 酵母提取物5,NaCl 10;pH 调为7.2(固体培养基添加2 g/dL 琼脂粉)。
TB 培养基(g/L):胰蛋白胨12,酵母提取物24,甘油6,KH2PO4,2.31,K2HPO412.54。
1.2 方法
1.2.1 目的基因的获得
1)C4ST-1 的获得 在NCBI 数据库检索来源于小鼠的C4ST-1 (NP_067414) 氨基酸序列,经TMHMM 软件预测分析C4ST-1 的第17-37 位氨基酸区域为跨膜区, 因此第38-352 位氨基酸所对应的基因序列(GenBank:AAI37630.1)按照E. coli密码子偏好性由南京金斯瑞生物科技有限公司进行全基因合成。
2)Erv1p的获得 采用酵母基因组提取试剂盒提取S. cerevisiaeS288C 基因组。 以S. cerevisiaeS288C 基因组为模板,设计引物F1 和R1,PCR 扩增获得Erv1p(GenBank:DAA08125.1)。
3)DsbC的获得 采用细菌基因组提取试剂盒提取E. coliBL21 (DE3) 基因组。 以E. coliBL21(DE3)基因组为模版,设计引物F2 和R2,PCR 扩增获得去除信号肽编码序列的DsbC(GenBank:CAQ33205.1)。
1.2.2 重组载体的构建
1)pET-32a(+)-C4ST-1 的构建 设计引物F3和R3 将质粒pET-32a(+)线性化。 设计与线性化质粒pET-32a(+)含有同源臂的引物F4 和R4,以合成的C4ST-1 为模板,PCR 扩增得到C4ST-1 产物。 将两步所得产物进行同源重组,转化E. coliJM109 感受态,挑取转化子测序,测序正确即获得重组质粒pET-32a(+)-C4ST-1,见图1。

图1 质粒pET32a(+)-C4ST-1 构建过程Fig. 1 Schematic of the construction of plasid pET32a(+)-C4ST-1
2)pRSFDuet1-Erv1p的构建 设计引物F5 和R5 将质粒pRSFDuet1 线性化。 将PCR 扩增获得的Erv1p片段与线性化质粒pRSFDuet1 进行同源重组,转化E. coliJM109 感受态,挑取转化子,测序正确则重组质粒pRSFDuet1-Erv1p构建成功,构建过程见图2(a)。
3)pRSFDuet1-DsbC的构建 设计引物F6 和R6 将质粒pRSFDuet1 线性化。 将PCR 扩增获得的DsbC与线性化质粒pRSFDuet1 进行同源重组,转化E. coliJM109 感受态,挑取转化子,测序正确则重组质粒pRSFDuet1-DsbC构建成功, 构建过程见图2(b)。
4)pRSFDuet1-Erv1p-DsbC的构建 利用引物F7 和R5 将质粒pRSFDuet1-DsbC线性化。 用引物F1 和R7 进行PCR 扩增得到Erv1p与线性化载体pRSFDuet1-DsbC进行同源重组。 转化E. coliJM109 感受态,挑取转化子测序,测序正确获得重组质粒pRSFDuet1-Erv1p-DsbC,构建过程见图2(c)。本文中所用引物均列于表1 中。

图2 表达载体pRSFDuet1-Erv1p、pRSFDuet1-DsbC、pRSFDuet1-Erv1p-DsbC 的构建Fig. 2 Construction of pRSFDuet1-Erv1p,pRSFDuet1-DsbC and pRSFDuet1-Erv1p-DsbC
1.2.3 重组载体的转化与诱导表达挑取测序正确的重组质粒转化E. coliBL21(DE3),挑取单菌落接种于装有3 mL LB 液体培养基 (含有50 μg/mL氨苄青霉素或卡那霉素)的12 mL 摇菌管中,37 ℃、220 r/min 过夜培养。 种子液按体积分数2%接种量转接至含50 mL TB (含有50 μg/mL 氨苄青霉素或卡那霉素)的250 mL 三角瓶中,37 ℃、220 r/min 培养至OD6000.6~0.8 左右, 添加终浓度为0.5 mmol/L的IPTG 诱导,25 ℃诱导表达16 h,收集菌体。

表1 PCR 扩增引物Table 1 Primers for PCR amplification
1.2.4 重组载体胞内粗酶液制备及其SDS-PAGE检测
1) 粗酶液制备 发酵结束后,将诱导表达产物于4 ℃、7000 r/min 离心10 min,弃上清液。 用pH 7.0的20 mmol/L Tris-HCl 将菌体洗涤2次, 稀释至OD6002.0~3.0,在冰上超声破碎(功率300 W,工作4 s,间歇6 s,10 min)。 4 ℃、12000 r/min 离心30 min,分别收集上清液与沉淀。所得上清液即为粗酶液,沉淀用与上清液同体积的pH 7.0 的20 mmol/L Tris-HCl 重悬,备用。
2) SDS-PAGE 检测 取30 μL 待检测样品,加入10 μL 的4×上样缓冲液混匀,于72 ℃变性10 min。取20 μL 变性样上样,120 V 开始电泳,当溴酚蓝指示剂迁移到距底部1~2 cm 处停止电泳。 电泳结束后, 取出胶块, 使用质量分数0.1%的考马斯亮蓝R250 染色液染色30 min 后纯水脱色, 至背景颜色较浅,凝胶成像分析仪拍照备用。
1.2.5 C4ST-1 的酶活性测定参照本课题组前期工作[12],向1 mL 20 mmol/L 的Tris-HCl(pH 7.0)反应缓冲体系中添加50 mmol/L PNPS,0.5 mmol/L PAP,2 mg ASST IV ,100 mg 软骨素,粗酶液2 mg,于37 ℃反应2 h。 以不加底物或将粗酶液加热失活作为阴性对照,沸水浴100 ℃、5 min 终止反应。 酶活力单位的定义为: 在最适反应条件 (37 ℃,pH 7.0)下,1 h 内催化生成1 μmol/L PNP 所需的酶量。
2 结果与分析
2.1 目的基因的扩增和重组载体的构建
2.1.1 pET-32a (+)-C4ST-1 的构建以合成的C4ST-1 为模板, 引物F3 和R3 进行PCR 扩增,扩增得到大约900 bp 的片段,见图3(a)。 采用DNA纯化回收试剂盒对单一凝胶片段进行回收,回收后的C4ST-1 片段与用引物F4 和R4 扩增得到的线性质粒pET-32a (+)(图略) 同源重组, 转化E. coli JM109, 挑取转化子交由上海生工生物工程有限公司测序, 测序结果正确, 重组载体pET-32a (+)-C4ST-1 构建成功。
2.1.2 pRSFDuet1-Erv1p 的构建以S. cerevisiae S288C 基因组为模板,引物F1 和R1 进行PCR 扩增获得约570 bp 的Erv1p 片段,见图3(b)。 将回收的Erv1p 片段和用引物F5 和R5 进行PCR 扩增得到的线性化载体pRSFDuet1(图略)进行同源重组,转化E. coli JM109,挑取转化子测序,测序结果显示正确,重组载体pRSFDuet1-Erv1p 构建成功。
2.1.3 pRSFDuet1-DsbC 的构建以E. coli BL21(DE3)基因组为模版,设计引物F2 和R2,PCR 扩增获得去除信号肽的约600 bp 的DsbC 片段,见图3(c)。 将回收的DsbC 片段与引物F6 和R6 进行PCR 扩增得到的线性化载体pRSFDuet1(图略)进行同源重组,转化E. coli JM109 菌株,挑取转化子测序,测序结果显示正确,重组载体pRSFDuet1-DsbC构建成功。

图3 PCR 产物的琼脂糖凝胶电泳分析Fig. 3 Analysis of PCR products by agarose gel electrophoresis
2.1.4 pRSFDuet1-Erv1p-DsbC 的构建设计引物F7 和R5 PCR 扩增得到线性化载体pRSFDuet1-DsbC (图略)。 用引物F1 和R7 进行PCR 扩增得到的Erv1p 片段(图略)与线性化载体pRSFDuet1-DsbC 同源重组,转化E. coli JM109 菌株,挑取转化子测序,测序结果显示正确。
2.2 C4ST-1 在E. coli 胞质表达的SDS-PAGE分析
将测序正确的重组质粒pET-32a (+)-C4ST-1转化E. coli BL21(DE3)感受态细胞,获得命名为E.coli BL21(DE3)- pET-32a(+)-C4ST-1 的宿主菌。以转化空质粒pET-32a(+)为对照。 挑取重组菌单菌落进行摇瓶水平发酵,发酵结束后对其胞内上清液及胞内沉淀进行SDS-PAGE 检测分析。结果如图4 所示,与对照相比,重组菌在沉淀的5.2×104处有一明显条带,上清中未观察到明显条带。 即C4ST-1在E. coli 胞质中主要以包涵体的形式存在。
2.3 共表达DsbC 对C4ST-1 的影响
以重组菌E. coli BL21 (DE3)- pET-32a (+)-C4ST-1 为出发菌株制备感受态细胞。 将重组质粒pRSFDuet1-DsbC 通过热击的方法转化至上述感受态细胞中,以转化空质粒pRSFDuet1 为对照。 重组菌经过摇瓶发酵, 超声破碎后进行SDS-PAGE 分析,结果见图5。 DsbC 重组蛋白在E. coli 中成功的进行了表达,其中DsbC(Mr=2.6×104)以胞内可溶形式表达(泳道3 蓝色箭头所示)。 C4ST-1 重组蛋白单独表达时主要以包涵体形式存在(泳道2),可溶部分未见明显条带 (泳道1); 共表达DsbC 导致C4ST-1 重组蛋白大部分以可溶形式存在 (第3 泳道),小部分以包涵体形式存在(第4 泳道)。 将各重组菌株进行摇瓶发酵,菌体超声破碎后所得上清液进行酶活测定, 结果见图6。 对照菌株的酶活为(9.42±0.29) U/L,DsbC 和C4ST-1 共表达菌株的酶活为(21.99±0.42) U/L,是对照菌株的2.33 倍。

图4 C4ST-1 表达的SDS-PAGE 分析Fig.4 SDS-PAGE result of expression product of C4ST-1
2.4 共表达Erv1p 对C4ST-1 的影响
C4ST-1 和Erv1p 共表达菌株的转化过程同2.3,将此菌株通过摇瓶发酵,超声破碎后进行SDSPAGE 分析,结果见图5。 Erv1p 在E. coli 中成功地进行了表达,Erv1p(Mr=2.63×104)以胞内可溶(泳道5 红色箭头所示)和不溶的2 种形式存在(泳道6 红色箭头所示)。共表达Erv1p 也能导致C4ST-1 融合蛋白部分以可溶形式存在(第5 泳道),但大部分仍以包涵体形式存在(第6 泳道)。即C4ST-1 与Erv1p共表达可在一定程度上促进C4ST-1 的可溶性表达,但其对C4ST-1 的可溶性表达影响相对较小。这可能因为Erv1p 为直接从酵母基因组中扩增得到,含有大量E. coli 稀有密码子, 在E. coli 中可溶性表达的量较少,故对C4ST-1 的促进作用有限。可根据E. coli 密码子偏好性将Erv1p 进行全基因合成,或以含有E. coli 稀有密码子的Rosetta(DE3)为宿主进行表达。 将C4ST-1 和Erv1p 共表达菌株进行酶活测定,结果见图6。 Erv1p 和C4ST-1 共表达菌株的酶活达到 (12.32±0.76) U/L, 是对照菌株的1.30 倍。
2.5 共表达Erv1p 和DsbC 对C4ST-1 的影响
C4ST-1、Erv1p 和DsbC 共表达菌株的转化 过程同2.3,将此菌株通过摇瓶发酵,超声破碎后进行SDS-PAGE 分析,结果见图5。 Erv1p 和DsbC 重组蛋白均在E. coli 中成功地进行了表达, 其中Erv1p(Mr=2.63×104)以胞内可溶(泳道7 红色箭头所示)和不溶 (泳道8 红色箭头所示) 的2 种形式存在;DsbC(Mr=2.6×104)以胞内可溶形式表达(泳道7 蓝色箭头所示)。 同时共表达C4ST-1、Erv1p 和DsbC导致C4ST-1 重组蛋白部分大部分以可溶形式存在(第7 泳道), 但沉淀仍见部分包涵体蛋白质条带(第8 泳道)。 将此重组菌株进行摇瓶发酵,菌体超声破碎后所得上清液进行酶活测定, 结果见图6。C4ST-1、Erv1p 和DsbC 共表达菌株的酶活比对照菌株增加2.09 倍,酶活最高可达(29.12±0.66) U/L。

图5 共表达Erv1p 和DsbC 对C4ST-1 表达的影响Fig. 5 Effect of co-expression of Erv1p and DsbC on C4ST-1

图6 共表达Erv1p 和DsbC 对C4ST-1 酶活的影响Fig. 6 Effect of co-expression of Erv1p and DsbC on C4ST-1 enzyme activity
2.6 C4ST-1、Erv1p 和DsbC 共表达菌株3 L 罐培养
为考察同时共表达C4ST-1、Erv1p 和DsbC 的菌株能否进一步工业化应用, 将此菌株进行3 L 罐发酵培养。 发酵过程控制转速250 r/min、溶氧水平在体积分数20%~30%、pH 7.0(流加氨水)左右、起始温度37 ℃,加入IPTG 后温度降低至25 ℃。 将保藏在甘油管的菌种接于LB 固体斜面培养基上进行活化,37 ℃培养过夜。挑取斜面菌种接入LB 液体培养基,37 ℃培养10 h 左右。 将种子液以体积分数为4%的接种量转接至装有1 L TB 培养基的3 L 发酵罐中,定时取样测定OD600和酶活,结果见图7。当菌体生长2 h 左右进入对数生长期, 加入终浓度为0.5 mmol/L 的IPTG 进行诱导表达。 当IPTG 诱导至10 h 左右菌体生长进入稳定期, 其酶活达到49.97 U/L。 继续延长培养时间,菌体生长不明显且酶活有所下降, 故10 h 左右为菌体的最佳产酶时间,为进一步的工业化应用提供技术支持。
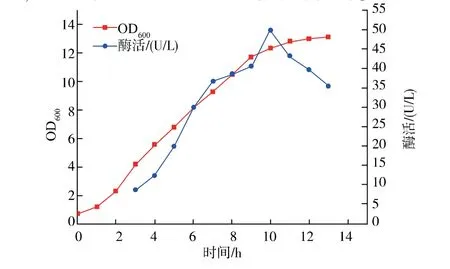
图7 3 L 发酵罐酶活变化曲线Fig. 7 Variation curve of enzyme activity under the fermentation condition
3 结 语
本研究中成功在E. coli 中表达了老鼠来源的C4ST-1, 但表达产物基本上以包涵体的形式存在,限制了其大规模应用。 C4ST-1 含7个不连续的半胱氨酸残基,当蛋白质含有2个以上的非连续二硫键时,其错误配对几率增大,异常的二硫化物需要异构化才能使蛋白质达到天然构象[27]。 为获得高可溶性表达和高活性的C4ST-1 重组蛋白, 本研究中共表达了参与二硫键从头形成的Erv1p 和促进二硫键正确配对和异构化的DsbC。 结果表明DsbC 和Erv1p 对C4ST-1 的可溶性表达量和酶活均有一定程度的提高。将C4ST-1、DsbC 和Erv1p 同时共表达的菌株进行3 L 发酵罐放大培养,10 h 左右酶活达到49.97 U/L,是原始菌株摇瓶的5.30 倍,对C4ST-1 的广泛应用奠定了一定的基础。 但本研究中对活性蛋白质的产生条件只进行了初步探索,重组蛋白仍有一大部分以包涵体的形式存在。 为进一步提高其可溶性表达量和酶活,一方面可以通过以胞质呈现氧化性环境的Shuffle 菌株[28-30]为宿主进行表达,另一方面可通过优化培养基、培养条件、构建不同的融合表达系统等进行尝试表达。
猜你喜欢
中国农业科学(2022年11期)2022-06-27
安徽农业科学(2022年1期)2022-02-14
江苏农业学报(2021年2期)2021-06-30
中等数学(2020年2期)2020-08-24
数学大王·低年级(2020年8期)2020-08-14
湖北农业科学(2017年10期)2017-06-22
科技与创新(2017年3期)2017-03-17
北京航空航天大学学报(2016年7期)2016-11-16
小雪花·初中高分作文(2016年9期)2016-05-14
知识窗(2011年5期)2011-05-14
