
慢粒灵制剂中青黛定性鉴别和红花3 种黄酮类成分含量测定*
2020-09-10 03:27赵金秀
中国药业 2020年17期
贾 勇,赵金秀
(河北省廊坊市中医医院,河北 廊坊 065000)
慢粒灵颗粒由三棱、莪术、青黛、山豆根、桃仁、红花、黄药子7 味中药组方,具有活血化瘀、散结消的作用,临床主要用于治疗瘀血阻滞证慢性粒细胞白血病[1]。方中三棱有消积止痛、破血行气作用[2];红花有散瘀止痛、活血通经作用,其活血成分为黄酮类成分[3-4],包括羟基红花黄色素A、山柰素、6-羟基山柰酚-3- O-β-葡萄糖苷和红花黄色素B 等。目前,质量标准中仅见青黛和山豆根的薄层色谱鉴别。青黛和山豆根主要是发挥清热解毒、消肿利咽的作用[5]。青黛中的靛玉红有治疗慢性粒细胞白血病作用[6],且根据中药质量标志物基于性-效-药(Q-Marker)的概念[7-8],中药质量标志物主要是指能与中药功能密切相关的成分,可作为反映中药安全性和有效性的标志性物质进行质量控制。因此,该质量标准的控制与中药质量标志物的概念关系不紧密,具有一定缺陷。本研究中对处方中青黛采用薄层色谱(TLC)法进行鉴别,采用超高效液相色谱(UPLC)法测定慢粒灵颗粒(胶囊)中羟基红花黄色素A、6-羟基山柰酚-3- O-β-葡萄糖苷和红花黄色素B,对其红花药材进行质量控制,可为提高和完善慢粒灵颗粒(胶囊)的质量标准提供参考。现报道如下。
1 仪器与试药
1.1 仪器
Water ACQUITY H-Class 型超高效液相色谱仪,包括Empower 3 工作站(美国沃特世仪器公司);Sartorius BT124 型电子天平(北京赛多利斯仪器公司,精度为万分之一);KQ-500DE 型数控超声波清洗器(江苏昆山超声仪器有限公司,功率为500 W,频率为40 kHz);Mili-Q 型超纯水机(美国密理博公司)。
1.2 试药
慢粒灵颗粒(医院制剂室自制,批号分别为20190320,20190706,20180516,规格为每袋15 g);慢粒灵胶囊( 医院制剂室自制,批号分别为20190704,20190730,20190801,规格为每粒0.45 g);羟基红花黄色素A 对照品(批号为111637-201308,含量为96.5%),靛蓝对照品(批号为110716-201612),靛玉红对照品(批号为110717-201805),购自中国食品药品检定研究院;6-羟基山柰酚-3- O-β-葡萄糖苷对照品(批号为MUST-19041506,含量99.72%),红花黄色素B 对照品(批号为MUST-19041507,含量99.68%),均购自成都曼斯特公司;乙腈(色谱纯,美国Fisher 公司);三氯甲烷、丙酮、甲苯、三氟乙酸、磷酸二氢铵、磷酸,均为分析纯,均购自上海国药集团化学试剂公司;水为超纯水。
2 方法与结果
2.1 青黛TLC 鉴别
取3 批慢粒灵颗粒各1 袋,研细,称取粉末各10 g;取胶囊20 粒,研细,分别置具塞锥形瓶中,加三氯甲烷20 mL,超声处理(功率为350 W,频率为50 kHz)10 min,滤过,滤液作为供试品溶液。另取靛蓝、靛玉红对照品,加氯仿制成每1 mL 各含0.5 mg 的溶液,作为对照品溶液。再取缺青黛的阴性样品,按供试品溶液制备方法制成阴性对照品溶液。照2015 年版《中国药典(四部)》通则0502 TLC 法试验,吸取上述4 种溶液各5 μL,分别点于同一硅胶G 薄层板上,以甲苯-三氯甲烷-丙酮(5 ∶4 ∶1,V/ V/ V)为展开剂,展开,取出,晾干,置日光下检视。供试品溶液色谱中,在与对照品溶液色谱相应位置上显相同颜色的斑点,阴性对照无干扰。色谱图见图1。

图1 薄层色谱图
2.2 含量测定
2.2.1 色谱条件
色 谱 柱:ACQUITY UPLC BEH C18柱(100 mm ×2.1 mm,1.7 μm);流动相:乙腈(A)-0.01%三氟乙酸水溶液(B),梯度洗脱,0~5.5 min 时2%A~5%A,5.5~8.4 min 时5%A~9%A,8.4~19.8 min 时,9%A~20%A,19.8~22 min 时20%A;流速:0.3 mL/min;柱温:30 ℃;检测波长:380 nm;进样量:10 μL。
2.2.2 溶液制备
取羟基红花黄色素A 对照品12.52 mg、6-羟基山柰酚-3- O-β-葡萄糖苷10.08 mg 和红花黄色素B 9.96 mg,精密称定,置同一25 mL 容量瓶中,加25%甲醇溶解并稀释至刻度,摇匀,即得混合对照品溶液。取慢粒灵颗粒(批号为20190320)10 袋,研细;取慢粒灵胶囊20 粒,研细。精密称取上述粉末各1.0 g 置150 mL 具塞锥形瓶中,分别加25% 甲醇50 mL,超声处理(功率为350 W,频率为50 kHz)30 min,取出,放冷,加25%甲醇补足减失的质量,摇匀,用微孔滤膜(0.45 μm)滤过,取续滤液,即得供试品溶液。取慢粒灵颗粒处方量的1 /10,制备缺红花药材的阴性样品,按供试品溶液制备方法制备红花阴性对照品溶液。
2.2.3 方法学考察
专属性试验:精密吸取上述3 种溶液各10 μL,注入液相色谱仪,按拟订色谱条件进样测定,记录色谱图。详见图2。可见,红花阴性样品对3 种成分的测定无干扰。
线性关系考察:取上述对照品贮备溶液作为5 号对照品溶液,分别精密吸取50,100,1 000,2 000 μL,分别置10 mL 容量瓶中,依次作为1~4 号对照品溶液。分别取上述1~5 号对照品溶液各10 μL,注入液相色谱仪,按拟订色谱条件进样测定,以进样量(X,ng)为横坐标、相对应峰面积(Y)为纵坐标进行线性回归。结果见表1。
精密度试验:分别精密收取线性关系考察项下的1号、3 号和5 号对照品溶液,按拟订色谱条件连续进样6 次。结果1 号对照品溶液中羟基红花黄色素A、6-羟基山柰酚-3- O-β-葡萄糖苷、红花黄色素B 峰面积的RSD 分别为1.98%,2.03%,1.65%(n=6);3 号对照品溶液中羟基红花黄色素A、6-羟基山柰酚-3-O-β-葡萄糖苷、红花黄色素B 峰面积的RSD 分别为1.37%,1.55%,1.12%(n=6);5 号对照品中羟基红花黄色素A、6-羟基山柰酚-3- O-β-葡萄糖苷、红花黄 色 素B 峰 面 积 的 RSD 分 别 为0.62% ,1.05% ,0.88%(n=6)。结果表明,仪器精密度良好。
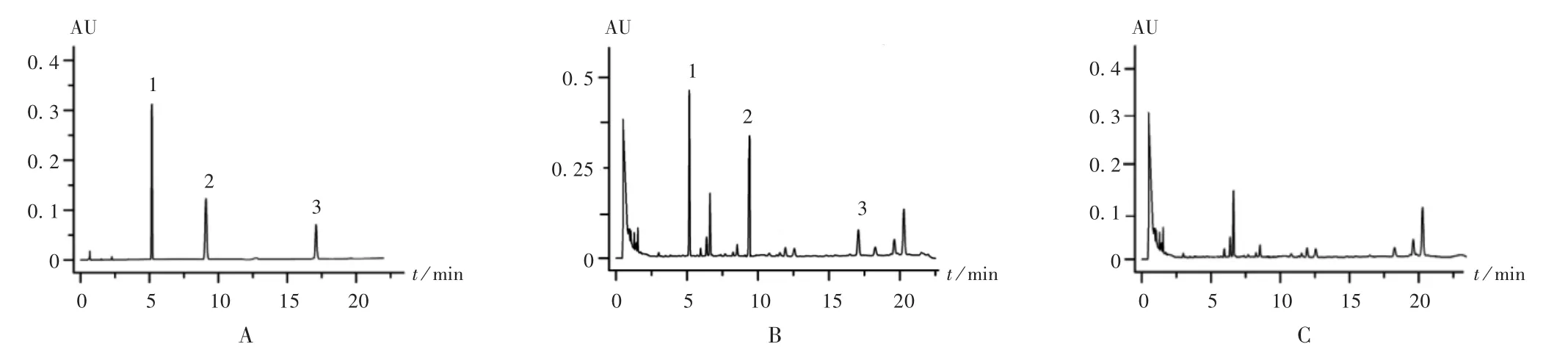
图2 慢粒灵颗粒中3 种黄酮类成分高效液相色谱图
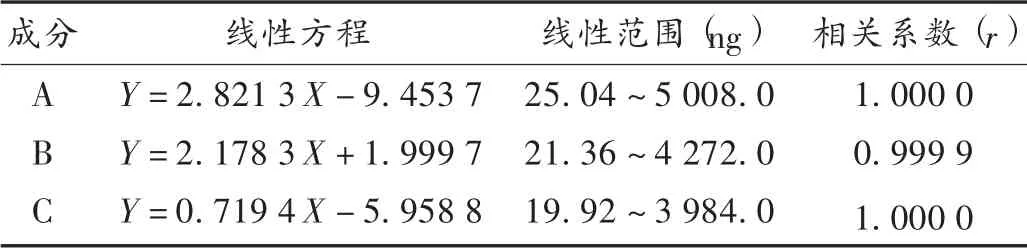
表1 3 种成分线性关系考察结果( n=3)
稳定性试验:取上述供试品溶液,分别于0,1,2,4,8,12,24 h 时按拟订色谱条件进样测定,连续考察24 h,记录色谱峰面积。结果羟基红花黄色素A、6-羟基山柰酚-3- O-β-葡萄糖苷和红花黄色素B 峰面积的RSD 分别为1.98%,2.32%,2.56%(n=6),表明供试品溶液在24 h 内稳定性良好。
重复性试验:取慢粒灵颗粒(批号为20190320),依法制备供试品溶液6 份,按拟订色谱条件分别进样测定。结果羟基红花黄色素A、6-羟基山柰酚-3- Oβ-葡萄糖苷和红花黄色素B 的平均含量分别为6.08,0.66,2.16mg/g,RSD 分别为2.01%,1.52%,1.36%(n=6),表明方法重复性良好。
加样回收试验:取羟基红花黄色素A 28.62 mg、6-羟基山柰酚-3- O-β-葡萄糖苷9.73 mg,精密称定,置同一10 mL 容量瓶中,加25%甲醇溶解稀释至刻度,作为待加溶液1;取红花黄色素B 8.28 mg,精密称定,置25 mL 容量瓶中,加25%甲醇溶解并稀释至刻度,作为待加溶液2;取已知含量的慢粒灵颗粒6 份,精密称定,每份0.5 g,置具塞锥形瓶中,向上述6 份样品中分别加入上述待加溶液1 和待加溶液2 各1 mL,依法制备供试品溶液,按拟订色谱条件进样测定,并计算回收率。结果见表2。
2.2.4 样品含量测定
取3 批慢粒灵颗粒/胶囊,依法制备供试品溶液,按拟订色谱条件进样测定,结果见表3。

表2 加样回收试验结果(n=6)

表3 慢粒灵颗粒/胶囊中3 种黄酮类成分含量测定结果(mg/g)
3 讨论
3.1 指标成分选择
预试验中,首先采用TLC 法对方中君药莪术和三棱进行薄层鉴别,结果发现二者相同成分较多,阴性成分互相有干扰,通过调整展开剂比例和不同的供试品制备方法均未能排除干扰,因此未将君药列入本研究。方中青黛的主要成分靛玉红具有消炎、抗肿瘤等药理活性,故选择TLC 法进行鉴别控制;红花有活血化瘀、消炎、抗肿瘤的功效,其中以黄酮类成分羟基红花黄色A、6-羟基山柰酚-3- O-β-葡萄糖苷和红花黄色素B 含量较高,因此,选择上述3 种成分为指标成分进行含量测定。
3.2 供试品溶液制备方法选择
首先考察不同提取溶剂(水、25%甲醇、75%甲醇和甲醇)的影响,结果发现甲醇作为提取溶剂时,羟基红花黄色A 和红花黄色素B 的含量下降较大;纯水作为溶剂时,色谱峰杂质较多,6-羟基山柰酚-3- O-β-葡萄糖苷峰形拖尾;25%甲醇作为提取溶剂时,3 种成分的含量相对较大,且峰形良好,分离度大于1.5。因此,选择25%甲醇作为提取溶剂。采用三因素三水平(制备方法为回流、超声、索氏提取;超声时间为20,30,40 min;超声功率为150,300,450 W)进行优化时,发现3 种提取方法提取率相差不大,超声功率对其也无明显影响,但随着超声时间的延长,含量逐渐增大,30 min 与40 min 含量接近。因此,确定供试品制备方法为25%甲醇超声(功率为300 W,频率为50 kHz),提取30 min。
3.3 测定条件选择
采用PDA 检测器对3 种成分进行全波长扫描,结果发现于380 nm 波长处3 种成分的干扰较小,峰形对称,分离效果好,故选择380 nm 作为3 种成分含量测定的最佳波长。由于黄酮类化合物易溶于甲醇、乙腈,因此流动相的选择先后采用甲醇-0.1%磷酸溶液、乙腈-0.1%甲酸水溶液、甲醇-0.5%乙酸溶液等多种流动相进行洗脱,结果发现,当乙腈-0.1%甲酸溶液作为流动相时,各色谱峰分离较好,峰形对称。故选择乙腈-0.1%甲酸水溶液作为流动相。
3.4 方法评价
本研究中建立的TLC 法专属性强,无干扰;UPLC法可同时测定红花中羟基红花黄色A、6-羟基山柰酚-3- O-β-葡萄糖苷和红花黄色素B 3 种黄酮类成分的含量,且具有分析速度快、准确性高、重复性好的优点,可用于慢粒灵颗粒中青黛和红花药材的质量控制,同时为提高慢粒灵颗粒(胶囊)的质量标准提供参考。
猜你喜欢
云南化工(2022年7期)2022-07-31
中西医结合心脑血管病杂志(2022年12期)2022-07-07
现代农村科技(2022年2期)2022-03-04
快乐语文(2021年34期)2022-01-18
快乐语文(2021年27期)2021-11-24
快乐语文(2021年11期)2021-07-20
快乐语文(2021年15期)2021-06-15
昆明医科大学学报(2021年1期)2021-02-07
中华养生保健(2020年1期)2020-11-16
人间(2015年11期)2016-01-09
