
原发性肺朗格汉斯细胞组织细胞增生症15例临床病理分析
2020-12-01 09:23王小燕郭芳芳徐紫光孔令非
临床与实验病理学杂志 2020年10期
王小燕,聂 宝,梅 昂,郭芳芳,徐紫光,成 琼,孔令非
肺朗格汉斯细胞组织细胞增生症(pulmonary Langerhans cell histiocytosis, PLCH)是朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis, LCH)的一个亚型,可分为两大类:一类为多系统LCH累及肺,多见于1~3岁儿童;另一类为少见原发性PLCH,而无其他脏器或系统受累,主要发生于成人,与吸烟高度相关,戒烟是首要的治疗手段[1]。原发性PLCH少见,国内外少有报道,因其临床表现、影像学改变与肺间质性疾病及肺癌相似,易误诊,确诊依赖肺活检[2]。本文回顾性分析15例原发性PLCH,探讨原发性PLCH的临床病理学特征、免疫表型、诊断、鉴别诊断及预后,并复习相关文献,旨在提高病理工作者对其的认识水平。
1 材料与方法
1.1 临床资料收集2010年1月~2019年8月河南省人民医院病理科诊断的15例原发性PLCH,所有病例均经两位专科病理医师复核确诊。影像学资料由本院放射科提供,查阅病历或电话直接咨询获取治疗及随访信息,随访时间截至2019年12月31日,患者失访或死亡,则随访终止。
1.2 方法
1.2.1HE及免疫组化 标本均经10%中性福尔马林固定,石蜡包埋,4 μm厚切片,行常规HE染色。免疫组化染色采用EnVision两步法,一抗包括S-100、CD1a、Langerin、CD68、CD3、CD20、Ki-67,均为即用型抗体,均购自丹麦Dako公司。
1.2.2PCR法 采用PCR法检测BRAF V600E基因突变。使用QIAamp DNA Micro Kit提取组织DNA,按人类BRAF基因突变检测试剂盒(厦门艾德生物公司)操作说明,采用PCR法判断基因是否突变。
2 结果
2.1 临床特征15例患者均为男性,年龄21~64岁,平均45岁。13例有吸烟史,所有患者均有咳嗽症状,同时伴胸痛(4/15)、咯血(2/15)、气促(3/15)、咳痰(4/15)、胸腔积液(1/15)、发热(1/15)。CT示双肺中上叶多发囊状透光影(图1)及少量边缘光滑的实性结节影,其中1例患者以双肺不均匀分布的软组织及团块影为主,周围见毛刺及条索影(图2),邻近胸膜局部增厚,考虑肺癌伴肺内多发转移。所有患者均行腹部CT及全身骨核素扫描检查,均未见病变受累。13例吸烟者首选戒烟治疗,其中9例同时行全身化疗(激素+蒽环类细胞毒药物),4例仅戒烟;另2例非吸烟患者,1例行单独激素治疗,1例行全身化疗。
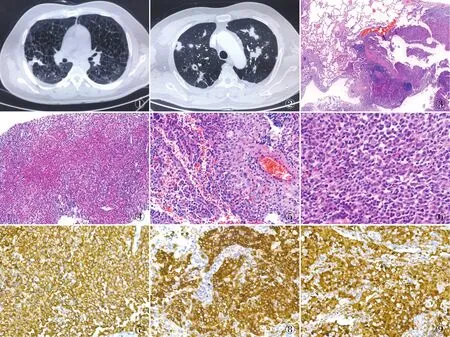
图1 CT示双肺多发囊状透光区 图2 CT示双肺不均匀分布软组织及团块影,周围见毛刺及条索影,部分病变呈分叶状,其内见空洞 图3 结节不均匀分布于细支气管周围、肺泡间隔或胸膜下 图4 大量朗格汉斯细胞、嗜酸性粒细胞、数量不等淋巴细胞、浆细胞及少量中性粒细胞形成的肉芽肿样结节,并见嗜酸性脓肿形成 图5 小血管壁内淋巴细胞、嗜酸性粒细胞浸润,管壁周大量朗格汉斯细胞增生聚集 图6 朗格汉斯细胞胞质丰富、淡嗜酸性,染色质细腻,可见核沟 图7 朗格汉斯细胞CD1a膜阳性,EnVision两步法 图8 朗格汉斯细胞Langerin胞质阳性,EnVision两步法 图9 朗格汉斯细胞S-100核质阳性,EnVision两步法
2.2 病理检查15例中5例行穿刺活检术,7例行楔形切除术,3例行肺段切除术。切除标本中,切面见肺组织呈多囊状改变。镜下见不均匀分布于细支气管周围、肺泡间隔或胸膜下的肉芽样结节,边界欠清(图3),结节内见大量朗格汉斯细胞、嗜酸性粒细胞、数量不等淋巴细胞、浆细胞及少量中性粒细胞浸润,并见嗜酸性脓肿形成(图4),部分结节中见囊性变,局灶小血管壁内淋巴细胞、嗜酸性粒细胞浸润,管壁周大量朗格汉斯细胞聚集(图5),高倍镜下朗格汉斯细胞胞界欠清,胞质丰富、淡染或嗜酸,核卵圆形、染色质细腻,可见核沟(图6)。切除标本中还可见不同区域的肺间质纤维组织增生伴慢性炎细胞浸润,肺泡腔内巨噬细胞聚集,朗格汉斯细胞数量较少,局灶可见瘢痕样改变及肺大泡形成。
2.3 免疫表型朗格汉斯细胞CD1a(15/15)(图7)、Langerin(12/15)(图8)、S-100(14/15)(图9)均阳性,CD68散在胞质阳性(3/15),Ki-67增殖指数5%~10%,结节内数量不等的淋巴细胞CD20或CD3阳性。
2.4 BRAF V600E基因突变情况15例中3例(3/15)BRAF V600E突变,其余12例均为野生型。
2.5 随访15例患者随访4~115个月。1例于诊断6个月后死于呼吸衰竭,截至随访结束,其余14例均生存良好。
3 讨论
LCH是一种局灶性、多灶性或播散性病变,常表现为朗格汉斯细胞克隆性增生,表达CD1a、Langerin和S-100,超微结构见Birbeck颗粒,目前认为其是一种炎性髓系肿瘤[3]。好发于儿童,每年发病率为0.02~0.09人/万,且男性略多,常累及骨、邻近软组织、皮肤、淋巴结等[4-5]。原发性PLCH是一种肺间质性病变,占弥漫性肺病变的3%~5%[6],90%患者为成年吸烟男性,病因不明,有学者认为与吸烟诱发的肺内过度免疫反应及长期慢性炎症所导致的肺间质小气道朗格汉斯细胞过度增生有关[7]。原发性PLCH与多系统LCH累及肺,两者病理改变类似,鉴别主要依靠临床特征,既往无LCH病史,影像学检查未发现其他部位/系统同时受累。
原发性PLCH一般起病隐匿,常见症状为咳嗽、呼吸困难、胸痛、咯血,CT示双肺结节、空洞及囊腔,以中上肺野为主,肋隔角区较轻[8]。本组15例均为成年男性,平均年龄45岁(21~64岁),13例有吸烟史,多以咳嗽为首发症状,CT示双肺多发囊腔,以双肺中上叶为主,肺肋隔角均未见病变,与文献报道基本一致,但本组中有1例患者以双肺不均匀分布的软组织及团块影为主,周围见毛刺及条索影,故考虑肺癌并双肺多发转移,活检组织病理诊断为LCH,提示LCH虽然有典型CT改变,但当出现较多结节伴毛刺影改变时,则与肺癌难以鉴别。腹部CT及全身骨核素扫描均未见病变受累,提示本组病例均为原发于肺的局限性LCH。原发性PLCH的治疗以戒烟为主,辅助皮质激素+蒽环类细胞毒药物的全身化疗[9]或靶向治疗[10],大多数成人PLCH患者预后较好[11]。本组患者采取戒烟和(或)全身化疗和(或)单激素治疗,截至随访结束,14例患者均生存良好,1例对全身化疗反应较差,于诊断后6个月死亡,该患者有长期吸烟、高血压及糖尿病史,咳嗽、咳痰症状多年,未予重视,一直按慢性支气管炎治疗,肺功能差,辗转多家医院治疗效果不佳后就诊于我院,CT检测结果考虑肺癌伴肺内多发转移,行穿刺活检诊断为LCH,分析该患者预后差可能与未及早确诊并治疗有关。
PLCH根据病情进展,形态学可分为3期[12]。(1)富细胞期:大量朗格汉斯细胞浸润构成的肉芽肿样结节,并见数量不等的嗜酸性粒细胞、浆细胞、淋巴细胞及中性粒细胞浸润;(2)增生期:可出现肺间质纤维化伴慢性炎细胞浸润,肺泡上皮增生,肺泡内大量巨噬细胞浸润,朗格汉斯细胞数量减少;(3)纤维化期:表现为较多瘢痕,无朗格汉斯细胞,间质可有纤维化、肺气肿、大泡形成。病变可以其中一种为主,也可混杂共存于同一病例,朗格汉斯细胞特异性表达Langerin、CD1a、S-100,故目前识别朗格汉斯细胞已不再依赖电镜下观察胞质内的Birbeck颗粒,本组Langerin(12/15)、CD1a(15/15)、S-100(14/15)阳性,与文献报道一致,仅病理形态、免疫表型与多系统LCH累及肺高度相似,故诊断原发性PLCH需结合临床、影像学检查。本组均可见富细胞期及不同程度的增生期改变,活检标本中仅见富细胞期改变,切除标本中可观察到富细胞期及增生期的过渡,灶区可见纤维瘢痕及肺大泡形成,提示本组病变病理分期较早,故而预后较好。本组1例患者仅行穿刺活检,获得的少量组织中仅观察到富细胞期改变,此形态难以解释患者的不良临床预后,这可能与活检的局限性有关。
目前,多项研究指出RAS-RAF-MEK-ERK通路在LCH发生、发展中发挥重要作用,LCH患者中BRAF V600E基因突变率为38%~64%,提示LCH可能属于肿瘤性病变[13-14]。而PLCH的BRAF V600E基因突变率约25%[15],且与患者预后不良、治疗疗效差有关[16],本组仅3例发生BRAF V600E基因突变,略低于文献报道,可能与本组病例数少有关。本组死亡患者中检出BRAF V600E基因突变,可能也提示与其差的临床预后有关。然而BRAF V600E基因突变与PLCH的发生、发展及预后的相关性,仍需大宗病例进一步明确。
原发性PLCH需与以下几种病变鉴别:(1)结节病及结核:结节病为双肺多发孤立结节而无囊腔形成,镜下表现为多发大小一致的肉芽肿,境界清,多沿支气管分布,肉芽肿内无坏死及朗格汉斯细胞、嗜酸性粒细胞增生,血清血管紧张素转换酶多增高。结核镜下见由上皮样细胞和(或)朗汉斯巨细胞构成的肉芽肿,中心常见凝固性坏死,结合抗酸染色及结核杆菌核酸检测,可明确诊断。(2)肺淋巴管平滑肌瘤病:虽然本病CT示双肺均匀分布囊腔,囊壁间见结节影,似PLCH,但几乎均发生于生育期女性,男性罕见,镜下见肺间质多灶分布胖梭形平滑肌细胞结节状增生可伴囊性变及少量淋巴细胞浸润。(3)呼吸性细支气管炎及脱屑性间质性肺炎:两者均是吸烟相关的双肺弥漫性间质性病变,临床表现及CT改变与PLCH相似,呼吸性细支气管炎表现为呼吸性细支气管、周围肺泡腔内见大量含棕色色素的组织细胞沉积,脱屑性间质性肺炎主要表现为肺泡腔内大量组织细胞沉积,两者均未见大量朗格汉斯细胞浸润。(4)特发性肺间质纤维化:与PLCH的纤维化期难鉴别,切除标本需广泛取材,仔细寻找是否存在由朗格汉斯细胞构成的肉芽肿样区域。(5)肺癌及转移癌:临床表现及CT改变与原发性PLCH可相似,但形态学完全不同,两者鉴别依赖于病理诊断。(6)Erdhein-Chester病:一种少见的黄色肉芽肿性组织细胞增生症,为多系统侵犯的疾病,累及肺时表现为沿脏层胸膜、支气管血管束和小叶见分布的大量泡沫样组织细胞浸润,可见杜顿巨细胞、淋巴细胞、浆细胞及嗜酸性粒细胞,CD68、CD163弥漫阳性,S-100少量阳性,但CD1a、Langerin阴性。
原发性PLCH好发于成年吸烟男性,其病理改变与多系统LCH累及肺无法鉴别,需结合临床及影像学特征,但明确诊断需经病理检查,以排除其他与PLCH临床表现及影像学改变相似的良、恶性病变。只要及时诊断并治疗,大多数原发性PLCH患者预后均较好。
猜你喜欢
家教世界·创新阅读(2022年4期)2022-05-07
情感读本·道德篇(2021年7期)2021-12-14
风流一代·经典文摘(2021年10期)2021-10-25
中国生殖健康(2020年2期)2021-01-18
国际放射医学核医学杂志(2020年2期)2020-05-30
优雅(2019年10期)2019-11-17
小学生导刊(2018年13期)2018-06-29
中国生殖健康(2018年2期)2018-01-12
小品文选刊(2016年1期)2016-02-12
中国神经精神疾病杂志(2014年1期)2014-03-01
