
同位素稀释-超高效液相色谱-串联质谱法快速同时测定胶原蛋白食品中16种激素
2020-12-07 10:04黄碧慧
现代食品·上 2020年10期
黄碧慧
摘 要:本文建立了一种基于超高效液相色谱-串联质谱(UPLC-MS/MS)同时测定胶原蛋白产品中激素的方法。样品用乙酸乙酯提取,用20%甲醇水进行转换溶剂后,用Plexa柱固相萃取柱净化,甲醇洗脱。以甲醇-水为流动相,样品经Kinetex C18柱(150 mm×3 mm,2.6 μm)梯度洗脱分离,在多反应监测模式下进行定性与定量分析。其中孕激素采用正离子扫描方式,雌激素采用负离子扫描方式,内标法定量,方法检出限为0.4~2.0 μg·kg-1。16种激素在高、中、低3个添加水平下的平均回收率分别为75%~115%,85%~105%,85%~118%,相对标准偏差分别为3.8%~13.4%、2.3%~10.4%和5.2%~14.0%。该方法操作简单、灵敏度高,适用于胶原蛋白产品中16种激素的检测。
关键词:超高效液相色谱-串联质谱;固相萃取;激素;胶原蛋白
Abstract:An UPLC-MS/MS based method for simultaneous determination of estrogen and progesterone in collagen products was developed. Sample was extracted by ethyl acetate, followed by solvent exchange and purification using Plexa SPE column. Further separation of the 16 target compounds was achieved on a Kinetex C18 column (150 mm×3 mm, 2.6 μm) via UPLC by gradient elution with using methanol/water as mobile phase. The analytes were determined qualitatively and quantitatively under the multi-reaction monitoring scan type with tandem mass analyzer, including a positive polarity mode for the progesterone, and a negative polarity mode for the estrogen, respectively. The analysis was performed using internal standards with detection limit of 0.4~ 2.0 μg·kg-1. The recovery of 16 hormones at low, median and high level is 75%~115%, 85%~105% and 85%~118% with relative standard deviation of 3.8%~13.4%, 2.3%~10.4% and 5.2%~14.0%, respectively. The method is simple and sensitive, and can be used for the detection of 16 hormones in collagen products.
Key words:UPLC-MS/MS; SPE; Hormones; Collagen foods
中圖分类号:O657.63
胶原蛋白在细胞再生,保证皮肤、骨骼发挥正常功能中有着重要的作用[1-2]。近年来,胶原蛋白类产品在市场上备受追捧,消费者购买需求旺盛,但胶原蛋白的一些负面报道也随之而来,由于一些激素有防止皮肤老化、除皱、增加皮肤弹性等作用[3-4],产品中非法添加激素的负面消息却不绝于耳。若长期食用这些激素,可对人体产生内分泌干扰作用,并影响身体健康。目前,市面上大部分胶原蛋白产品属于食品中的饮料类、糖果类等,这类食品的监管很少涉及激素项目的检测,并且也暂无关于这类食品中多激素检测的相关方法标准,因此建立一种胶原蛋白产品中多激素的检测方法显得十分必要。
胶原蛋白产品中含丰富的蛋白、糖等基质,在检测过程中必然存在基质效应(ME)的问题,增加了多激素净化和检测的难度。目前,多激素检测普遍采用的方法有气质联用法[5-7]、液相色谱法[8-10]和液质联用法[11-15],其中液质联用法因其高稳定、特异性强、前处理简单不需要衍生等特点,近年来被广泛应用于各种化合物的检测中。本研究建立了同位素内标-高效液相色谱-串联质谱仪胶原蛋白产品中7种雌激素(雌三醇、雌二醇、炔雌醇、雌酮、己烯雌酚、己烷雌酚和己二烯雌酚)、9种孕激素(炔诺酮、21α-羟基孕酮、17α-羟基孕酮、甲基炔诺酮、甲羟孕酮、乙酸甲地孕酮、乙酸氯地孕酮、孕酮和甲羟孕酮乙酸酯)的测定分析方法,实现对各类激素的快速准确测定。
1 材料与方法
1.1 仪器设备、试剂与耗材
API 5500Q Trap型三重四级杆质谱仪(美国AB公司);1290超高压液相色谱分离系统(美国安捷伦公司);20通道固相萃取装置(美国安捷伦公司);MS2旋涡混匀器(德国IKA有限公司);CF15RN高速冷冻离心机(日本Hitachi公司);MV5氮吹浓缩仪(美国LabTech公司)。
标准品购自德国Dr. Ehrenstorfe,包括雌三醇、雌二醇、炔雌醇、雌酮、己烯雌酚、己烷雌酚、己二烯雌酚、炔诺酮、21α-羟基孕酮、17α-羟基孕酮、甲基炔诺酮、甲羟孕酮、乙酸甲地孕酮、乙酸氯地孕酮、孕酮和甲羟孕酮乙酸酯(纯度>97%)共16种激素。以甲醇为溶剂配制成质量浓度为100 mg·L-1的单标储备液,于-20 ℃储存。临用时根据需要配制所需的浓度。
11种激素内标,分别为炔诺孕酮-D6、孕酮-D9、甲地孕酮乙酸酯-D3、甲羟孕酮-D3、美仑孕酮-D2、炔诺酮-13C2、雌酮-D2、雌二醇-13C2、己烯雌酚-D8、己二烯雌酚-D2和己烷雌酚-D4(纯度>97%)。以甲醇为溶剂配制成质量浓度为10 mg·L-1的单标储备液,于-20 ℃储存。
乙酸乙酯、甲醇,均为色谱纯,购自美国默克公司;实验用水为自制Mill-Q超纯水。
固相萃取柱:Bond Elut Plexa柱(3 mL/60 mg,Agilent公司)。液相色谱柱Kinetex C18(150 mm×3 mm, 2.6 μm,美国Phenomenex公司)。
实验用的胶原蛋白类食品于网络上采购收集,包括胶原蛋白糖果、胶原蛋白固体饮料和胶原蛋白饮品3种类型产品。
1.2 样品前处理
1.2.1 样品的制备
取多个预包装的样品直接混合均匀,胶原蛋白糖果样品充分粉碎并搅拌均匀。取其中的200 g装入玻璃容器中,密封,试样于4 ℃保存。
1.2.2 样品的提取
称取(2±0.01)g制备后的样品于50 mL聚乙烯离心管中,其中糖果与固体饮料需加入少量水(视情况而定)至样品溶解,而液体饮品不需要加水,然后加入5 mL乙酸乙酯溶液,2 000 r·min-1涡旋混合1 min,在4 ℃,12 000 r·min-1离心3 min,重复提取2次,合并上清液。
1.2.3 样品的净化
将上清液氮吹至近干后,加入2 mL,20%甲醇水(v∶v)复溶,涡旋混匀,待净化过柱。加入依次以6 mL甲醇和6 mL水活化的Plexa小柱,以1滴/s的速度過柱,弃去流出液,再用6 mL 25%的甲醇水(v∶v)进行淋洗,抽干,最后用8 mL甲醇进行洗脱,收集洗脱液在40℃氮吹浓缩至近干,用1.0 mL,20%甲醇水(v∶v)溶液复溶,涡旋混匀,过0.22 μm滤膜,上机待测。
1.3 分析条件
1.3.1 色谱条件
柱温:35 ℃,流速0.3 mL·min-1,进样量:2.0 μL。流动相:10%甲醇水(v:v)(A1)和甲醇(B1);梯度洗脱程序:0~1 min,40% B1;1~6 min,40%~60% B1;6~9 min,60%~90% B1;9~10 min,90% B1;10.1~15 min,40% B1。
1.3.2 质谱条件
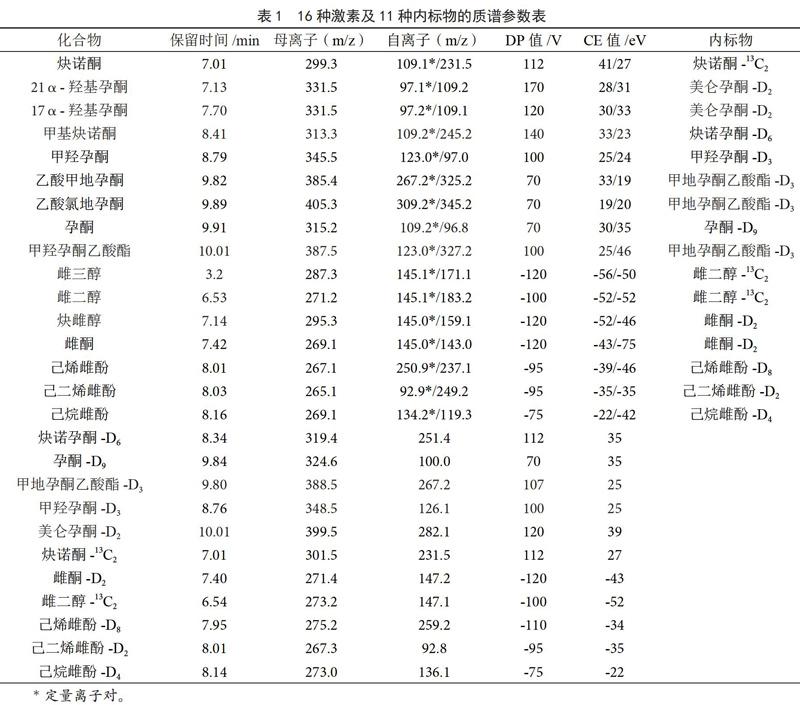
离子源:电喷雾电离源;扫描方式:正负离子同时监测;气帘气压(CUR):25 MPa;电喷雾电压(IS):5 500 V;离子化温度(TEM):550 ℃;雾化气压力(GS1):50 MPa;干燥气压力(GS2):50 MPa。目标物的质谱参数定性、定量离子对,去簇电压(DP)及碰撞电压(CE)见表1。
2 结果与分析
2.1 前处理方法优化
2.1.1 提取溶剂的选择
实验中的化合物属于非极性物质,根据相似相溶的原理,提取溶剂也应选用非极性溶剂,乙酸乙酯作为非极性物质提取溶剂有着不可取代的优势,尤其在处理含有大量水分的液体样品,与乙腈、甲醇等与水能互溶的提取溶剂相比,前处理还能减少繁琐的除去样品中水分的步骤,实现快速提取测定的目的,因此本文选择乙酸乙酯作为实验的提取溶剂。另外,由于固体样品不溶于乙酸乙酯,为了使样品提取更完全,在处理固体样品时,先加少部分水,将样品溶解后,再加适量的乙酸乙酯提取。
2.1.2 净化方法的优化
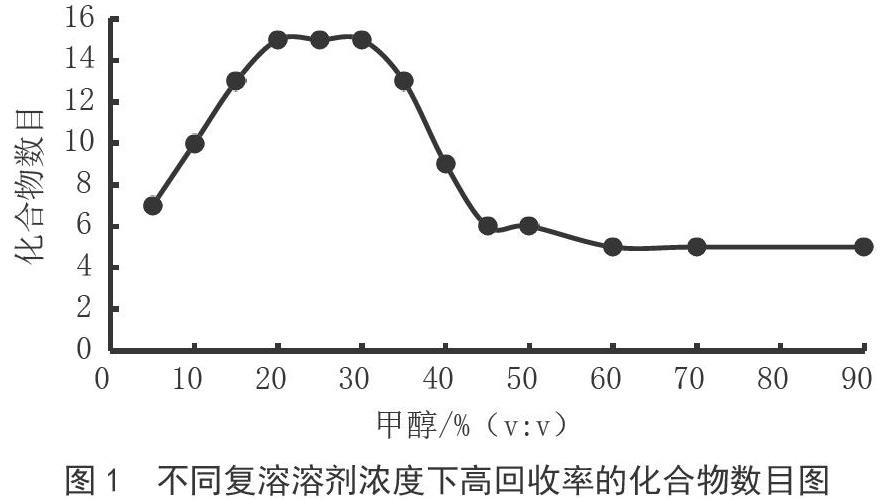
16种化合物结构中没有铵盐、羧酸官能团,因此不适宜选择用离子交换柱作为净化样品的固相萃取柱。Plexa柱是一种非极性二乙烯基苯基的中性聚合物吸附剂,可以利用其非极性保留机理净化样品。同时为了在上样过程中能更好地在柱上有保留,在用乙酸乙酯提取完成后,要先进行溶剂转换。复溶溶剂为一定浓度的甲醇水溶液,复溶溶剂中甲醇的含量不能过高,过高会导致上样时目标化合物在固相萃取柱的不保留,过低不利于非极性目标物的溶解。本实验考察了不同体积分数比的甲醇水溶液作为上样溶剂时的处理效果。不同复溶溶剂浓度下高回收率的化合物数目如图1所示,结果表明甲醇体积比在20%时,所有目标化合物的回收率能达到90%以上。
2.2 色谱条件的优化
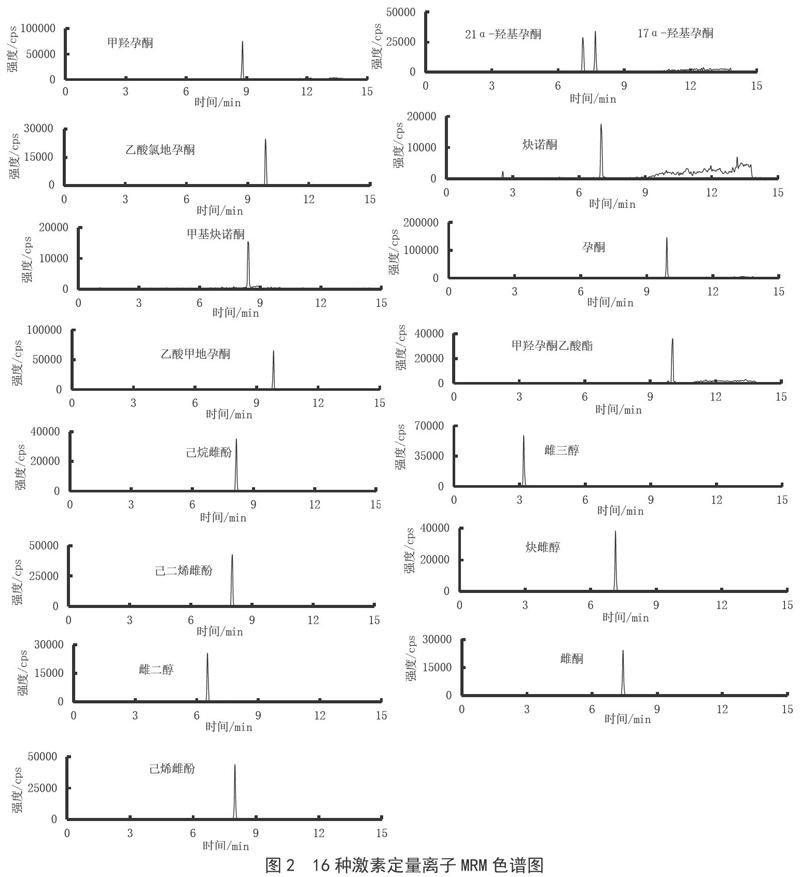
本研究采用了Phenomenex的Kinetex C18液相色谱柱,能有效分离16种激素化合物。质谱采集采用了正负离子同时监测的MRM模式,因此在选择流动相体系时,应同时兼顾正负离子模式下的离子化效率,让各化合物都能达到良好的响应。本研究考察了在乙腈-0.1%甲酸水、乙腈-5 mmol·L-1乙酸铵、甲醇-水3种流动相体系下目标化合物的灵敏度和重现性,结果表明:正离子采集模式下的孕激素在乙腈-0.1%甲酸水和甲醇-水流动相体系下的响应值接近,都比在乙腈-5 mmol·L-1乙酸铵体系下的响应值高。但孕激素在乙腈-0.1%甲酸水体系下的噪音强,基线高;在另外两个体系下的基线低,性噪比高。负离子采集模式下的雌激素在乙腈-0.1%甲酸水体系下雌三醇、雌二醇、炔雌醇、雌酮无响应,其余化合物响应也不高;在乙腈-5 mmol·L-1乙酸铵体系下响应次之;在甲醇-水体系中的响应最高,峰形好、性噪比最高。综合上述实验结果,最终选择甲醇-水为分析流动相。16种激素定量离子MRM色谱图见图2。
2.3 质谱条件的优化
本研究采用大气压电喷雾离子源,正负离子同时监测模式对16种激素进行测定,采用蠕动泵进样方式,将16种激素标准溶液(其中炔雌醇、雌二醇、雌三醇浓度为200 ng·mL-1,其余各激素为100 ng·mL-1)分别注入质谱中。先进行一级质谱分析,得到各自的母离子。再对目标化合物进行二级质谱扫描,得到物质的子离子信息,最后通过优化各物质的去簇电压(DP)和碰撞能量(CE),确定各目标化合物的质谱参数,质谱参数见表1。
2.4 方法学评价
2.4.1 基质效应
基质效应在质谱法中普遍存在,影响实验的准确度和精密度。因此在高效液相色谱-质谱分析方法开发和验证过程中需要对基质效应进行评价[16]。本文分别采用甲醇和胶原蛋白饮品空白基质液配制激素的混标溶液,其中炔雌醇、雌二醇、雌三醇浓度为20 ng·mL-1,其余激素浓度为10 ng·mL-1。分别上机测定,比较在没有基质和有基质的条件下,各激素的响应值变化。基质效应的计算方法见公式(1)。
由测定结果可知,正离子模式采集下的孕激素基质效应不是很明显,只有炔诺酮和甲基炔诺酮有略微的抑制效应,抑制率在60%~70%。但负离子模式下采集的雌激素全都有基质增强效应,增强率在110%~150%。基质效应的存在,影响了目标物的测定结果。由于内标物与目标化合物有相同的化学性质,在质谱检测过程中能有相似的响应行为,因此采用内标法定量最能有效的消除基质效应。
2.4.2 内标物的选择
实验本着尽量选择与目标物相对应的同位素化合物作为内标物的原则,对16种性激素进行定量。但仍有部分化合物(如21α-羟基孕酮、17α-羟基孕酮、乙酸氯地孕酮、甲羟孕酮乙酸酯、炔雌醇、雌三醇)无相应的同位素化合物。因此,为了达到准确定量的目的,通过分析标准品与内标物的响应数据,选择出峰时间接近、并与目标化合物有相应响应行为的同位素化合物作为其定量采用的内标物。化合物对应的内标物见表1。
2.4.3 线性范围、检出限
本研究采用内标法对目标化合物进行定量分析。配制系列梯度水平的目标物溶液,内含相同浓度的内标液(除雌二醇-13C2浓度为10.0 ng·mL-1外,其余内标浓度为5.0 ng·mL-1)作为标准溶液。以定量离子峰面积为纵坐标,以各化合物浓度与相应的内标物浓度的比值为横坐标进行线性回归,以最低浓度响应的3 倍信噪比(S/N≥3)对应的浓度作为检出限(LOD),以10倍信噪比(S/N≥10)对应的浓度作为定量限(LOQ)。结果见表2。从表2可以看出,16种化合物在一定的范围内线性关系良好,其相关系数达到了0.99以上。
2.4.4 回收率和精密度
按照2.2节的前处理方法分别选取胶原蛋白饮品、胶原蛋白固体饮料、胶原蛋白糖果空白样品为测试对象,考察了3类样品高、中、低(2 μg·kg-1、10 μg·kg-1、20 μg·kg-1)3个水平的加标实验,每个添加水平平行测定6次。求出各化合物的平均回收率和相对标准偏差(relative standard deviation,RSD)。测定结果显示:在胶原蛋白飲品、胶原蛋白固体饮料、胶原蛋白糖果样品中,16种激素在2 μg·kg-1加标水平时,相对标准偏差为3.8%~13.4%,平均回收率为75%~115%;在10 μg·kg-1加标水平时,相对标准偏差为2.3%~10.4%,平均回收率为85%~105%;在20 μg·kg-1加标水平时,相对标准偏差为5.2%~14.0%,平均回收率为85%~118%。方法准确度和精密度均满足检测的要求。
2.5 实际样品检测
运用本研究建立的方法对市售胶原蛋白饮品、胶原蛋白固体饮料、胶原蛋白糖果10份样品进行16种激素的检测,检测结果显示未见有目标激素检出。
3 结论
本研究建立了同位素稀释-固相萃取-超高效液相色谱-串联质谱法同时测定胶原蛋白食品中16种激素的检测分析方法,该方法具有简单快捷、灵敏度高、准确度和精密度高等优点。使用乙酸乙酯作为提取溶剂,固相萃取法净化,同时利用同位素内标消除样品基质效应,使样品前处理方法适合不同类型的胶原蛋白产品。选择合适的正负离子同时采集的流动相体系,实现一次上机完成16种激素同时测定的目的。
参考文献:
[1]周雪松.胶原蛋白肽产业现状及发展趋势[J].食品与发酵工业,2013,39(6):111-115.
[2]Farndale R W. Collagen-binding proteins:insights from the Collagen Toolkits[J]. Essays In Biochemistry,2019,63(3):337-348.
[3]王丽,张伶俐.激素补充治疗的安全性研究进展[J].实用妇产科杂志,2016,32(4):262-265.
[4]袁丽君,徐庄剑,许祥生.牛奶中的雌激素及其安全性[J].国际内科学杂志,2007,34(4):712-714.
[5]孙汉文,李挥,康占省,等.气相色谱-质谱法同时检测婴幼儿配方奶粉中的6种雌激素残留[J].河北大学学报(自然科学版),2011,6(3):607-611.
[6]马丽莎,戴晓欣,谢文平,等.QuEChERS/GC-MS法同时测定鱼、虾中的雌二醇与己烯雌酚残留[J].分析测试学报,2015,34(1):62-66,72.
[7]Sena K,Dotse S C,Merve F,et al. Determination of endocrine disruptive phenolic compounds by gas chromatography mass spectrometry after multivariate optimization of switchable liquid-liquid microextraction and assessment of green profile[J].Chemosphere,2019,235:205-210.
[8]王全一,矫筱蔓,董宇,等.高效液相色谱法检测中成药中4种雌激素的方法研究[J].药物分析杂志,2011,31(2):344-347.
[9]程俊.高效液相色谱-二极管阵列/荧光检测器串联法同时测定化妆品中8种性激素的研究[J].分析试验室,2016,35(1):112-116.
[10]周江,黄艳美,龚越飞,等.超高效液相色谱法測定美白、祛痘化妆品中4种禁用激素[J].化学分析计量,2016,25(2):52-55.
[11]Mengting W,Yang W,Bin P,et al. Multi-class determination of steroid hormones and antibiotics in fatty hotpot ingredients by pressurized liquid extraction and liquid chromatography–tandem mass spectrometry[J]. Journal of Pharmaceutical and Biomedical Analysis,2019,171:193-203.
[12]杨金泉,贺小敏,施敏芳,等.固相萃取-超高效液相色谱-串联质谱法同时测定地表水中9种性激素[J].中国环境监测,2019(3):19-27.
[13]Aga D S,He P. Comparison of GC-MS/MS and LC-MS/MS for the analysis of hormones and pesticides in surface waters: Advantages and pitfalls[J].Analytical Methods,2019,11(11):1436-1448.
[14]冯月超,贾丽,王建凤,等.超高效液相色谱-质谱联用法检测鱼肉中23种性激素残留量[J].食品安全质量检测学报,2018,9(16):4417-4426.
[15]马微,程丽,张兰威,等.基质分散固相萃取-高效液相色谱-串联质谱法同时测定减肥保健食品中20种违禁添加药物[J].分析化学,2014,42(8):1161-1170.
[16]Liu Z C,Yang F,Yu K J,etal. Multi-residue determination of five antiviral drugs in chicken tissues by liquid chromatography-electrospray ionization tandem mass spectrometry[J].Chinese Journal of Chromatography,2012,30(12):1253-1259.
猜你喜欢
中国中药杂志(2016年21期)2017-02-16
中国中药杂志(2016年21期)2017-02-16
分析化学(2017年1期)2017-02-06
分析化学(2017年1期)2017-02-06
热带农业科学(2016年10期)2016-12-12
分析化学(2016年7期)2016-12-08
上海医药(2016年1期)2016-02-22
安徽农学通报(2015年10期)2015-06-15
肉类研究(2015年1期)2015-04-08
分析化学(2014年3期)2014-12-16
