
改进的动态PPI网络构建与蛋白质功能预测算法
2020-12-16 02:41罗爱静瞿昊宇许家祺
计算机工程 2020年12期
李 鹏,闵 慧,罗爱静,瞿昊宇,伊 娜,许家祺
(1.中南大学湘雅三医院,长沙 410006; 2.湖南中医药大学 信息科学与工程学院,长沙 410208; 3.医学信息研究湖南省普通高等学校重点实验室(中南大学),长沙 410006; 4.湖南信息职业技术学院 软件学院,长沙 410200)
0 概述
随着人类基因组计划以及多个物种全基因组测序工作的完成,目前生命科学研究的重点已经转变为蛋白组学[1]。蛋白质是指由多种氨基酸按照某一规律采用多肽键所构成的一种多分子化合物,其是生物体中细胞的重要成分,也是生物体完成生命活动最重要的物质基础[2]。一个生物体内所有蛋白质的相互作用构成了蛋白质相互作用网络(Protein-Protein Interaction Network,PPIN),简称蛋白质网络[3]。值得注意的是,蛋白质之间的相互作用是动态的,它会随着时间环境、蛋白质的存在和降解、细胞的不同生理状态等因素的变化而变化。但由于PPIN本身的复杂性、可利用蛋白质相互作用数据的不完全性和噪声等诸多因素,准确且高效地衡量蛋白质相互作用的动态性还存在很多挑战[4],这也直接限制了PPIN领域内其他问题(如复合物挖掘[5]、关键蛋白识别[6]、网络比对[7]等)的研究进展。
文献[8]从表达动态性、多状态下表达及相关性变化和时空动态变化3个角度讨论了动态蛋白质网络的构建问题,在此基础上介绍动态蛋白质网络在复合物识别、疾病基因检测等方面的应用,并指出未来动态蛋白质网络所面临的挑战。文献[9]考虑到酵母物种中蛋白质的基因表达具有时间周期性这一特性,将PPI网络数据和时间序列基因表达数据相结合构建动态蛋白质交互网络(Dynamic Protein Interaction Network,D-PIN),并提出一种蛋白质功能预测方法。该文主要通过基于时间的采样来构建D-PIN,但对不同物种而言,如何合理地选择一个合适的时机进行采样仍缺乏理论指导。文献[10]针对蛋白质功能标签数量庞大且标签关联性较高的特点,提出一种基于布尔矩阵分解的蛋白质功能预测框架PFP-BMD,然而该框架在降低数据噪声影响方面的效果欠佳。文献[11]提出一种基于多关系网络中关键功能模块挖掘的蛋白质功能预测算法PEFM。该算法以高内聚低耦合的原则寻找关键功能模块,并利用这些功能模块中的邻居蛋白质信息来注释未知蛋白质的功能。然而由于需要在多个关系网络中进行查找,一旦蛋白质之间的相互作用发生改变(如蛋白质降解),则预测效果直线下降,不适用于动态蛋白质网络。文献[12]针对现有蛋白质功能预测方法预测精度不高、易受数据噪声影响等问题,提出一种基于机器学习的蛋白质功能预测方法HPMM,主要采用层次聚类、主成分分析和多层感知器等技术来实现功能预测。然而该方法在训练多层感知器过程中需要估计的参数较多,时间复杂度较高,且仅适用于静态蛋白质网络。
针对以上方法的不足,本文对动态蛋白质网络的构建问题进行研究,基于进化图提出一种改进的动态蛋白质网络构建算法,在此基础上设计蛋白质功能预测算法IPA-PF,并通过仿真实验验证算法的有效性。
1 动态蛋白质网络构建算法
由于蛋白质之间的相互作用并不是一成不变的,因此本文采用进化图[13]对动态蛋白质网络进行建模。为便于描述,给出建模过程中用到的定义:
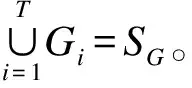
定义2(蛋白质的活性周期) 对于任意给定的一个蛋白质P,如果在一个给定的时间周期T内P的基因表达平均值u(P)都不低于阈值ε,则称T(P)为P的活性周期。
1.1 动态蛋白质网络的构建
根据上述定义,动态蛋白质网络的构建主要包含以下3个步骤:
步骤1根据蛋白质基因表达数据的平均值计算所有蛋白质的活性周期。
步骤2根据所有蛋白质的不同活性周期划分出多个时间片,拥有相同活性周期的蛋白质属于同一个时间片。对于处于同一时间片的所有蛋白质,根据它们之间的连接强度构成一个蛋白质子网。
步骤3对步骤2得到的各个时间片的子网,采用进化图进行建模,最终得到一个全局的动态蛋白质网络。
1.1.1 活性周期计算
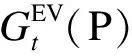
(1)
(2)
进一步地,本文采用F(P)反映蛋白质P基因表达曲线的波动性:
(3)
可以看出,标准差越大,F越小,F的范围为[0,1]。活性阈值ε的选取参考文献[14]中提出的3-sigma准则,如下所示:
ε=u(P)×F(P)+(u(P)+3σ(P))×(1-F(P))
(4)
如果在某一时间片Tx内有u(Pi)≥ε,i=1,2,…,k,则认为这k个蛋白质具有相同的活性周期,可用于构建同一个蛋白质子网。通过活性周期的计算可以得到一个关于所有蛋白质活性周期的集合S_T={T1,T2,…,Tk}。本文根据S_T中元素的个数决定划分出时间片的个数,即构建子网的个数。
1.1.2 蛋白质子网构建
以某一个子网为例来阐述其构建过程,其余子网的构建与此类似。设P_S={P1,P2,…,Pn}表示具有相同活性周期(同一时间片)的所有蛋白质集合,要在这n个蛋白质之间构造一个子网,即要找到n个蛋白质之间的相互作用关系。本文通过考查这些蛋白质之间的连接强度来判断它们之间是否具有相互作用,如果认为它们之间有相互作用,则在这两个蛋白质之间添加一条边。
连接强度主要从两方面衡量,即直接连接数和间接连接数。直接连接数主要是指两个蛋白质之间拥有的共同邻居节点数,如果两个蛋白质有更多共同邻居,则表明这两个节点之间的关系更为紧密,更有可能发生相互作用;间接连接数指两个蛋白质之间直接相连的边数和节点的度最小值的比值,它也可以用来衡量蛋白质之间相互作用的强弱。因此,连接强度的定义如下所示:
定义3(连接强度) 蛋白质Pi和蛋白质Pj之间的连接强度JS(Pi,Pj)计算公式如下:
(5)
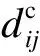
1.2 动态蛋白质网络模型
动态蛋白质网络与静态蛋白质网络的本质区别在于网络拓扑因时间、外界环境等因素的动态变化而导致连通性动态变化。如何利用合适的模型来刻画这种动态性是对蛋白质网络准确建模的关键。考虑到蛋白质的基因表达值具有时间周期性,本文首先将整个蛋白质网络的运行时间划分为多个时间片,刻画出每个时间片内的连通情况,然后利用进化图的时间演化特性将连续时间片内的多个子图构建为运行时间内的进化图模型。
图1给出了蛋白质网络工作过程中不同时刻节点相互作用的动态变化情况。其中,顶点是蛋白质,边表示蛋白质之间的相互作用。假设T1~T4为整个网络生命周期内任意4个连续的时间片,分别可以构建得到这4个连续时间片内的网络快照。

图1 动态蛋白质网络连续时间片快照Fig.1 Snapshots of continuous time slices ofdynamic protein network
根据定义1,将图1所示的连续时间片快照建模为进化图模型。图1所示时间片快照中的蛋白质(A,B,C,D,E,F,G,H,I,J,K,L)对应于定义1中的顶点集合V,边集合对应于定义1中的边集合E,时间序列集合(T1,T2,T3,T4)对应于定义1中的有序时间序列TS。建模过程如下:
1)构造T1时间片内蛋白质网络连通情况所对应的进化图子图G1,并在新出现的每条边上增加时间序列元素T1。
2)在G1的基础上累加构造T2时间片内蛋白质网络连通情况所对应的进化图子图G2,并在T2时间片内出现的边上增加时间序列元素T2。
3)以此类推,直到全部的时间片所对应的进化图子图构造完成,得到的进化图模型如图2所示。其中,每条边上的数字序列代表该相互作用存在对应的时间序列,标识该相互作用在第几个时间片中出现,例如蛋白质A和蛋白质D只在第1个、第2个和第4个时间片内存在相互作用。
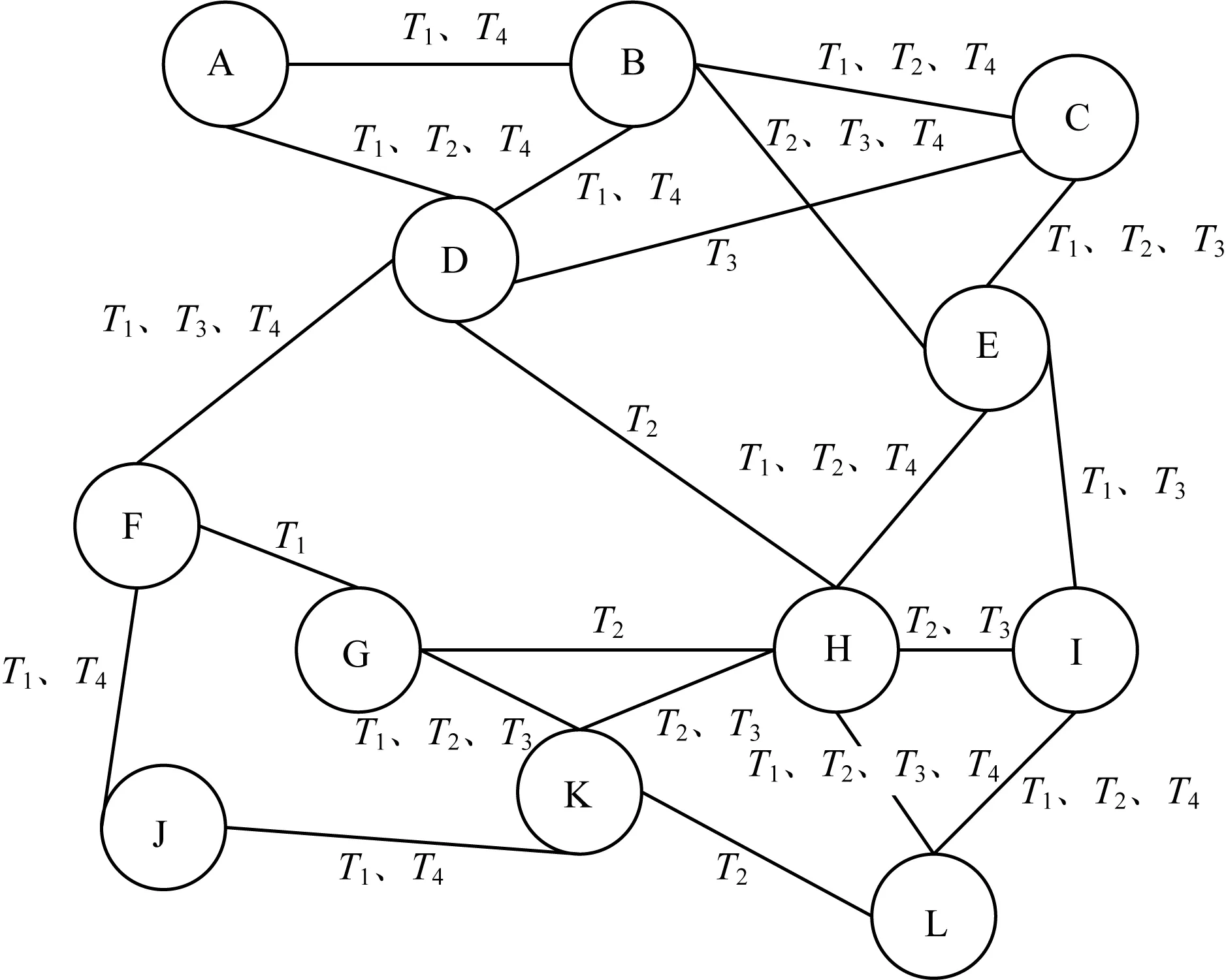
图2 基于进化图的动态蛋白质网络模型Fig.2 Dynamic protein network model based onevolutionary graph
本文提出的动态蛋白质网络构建算法描述如下:
算法1动态蛋白质网络构建算法
输入蛋白质相互作用数据,阈值th,基因表达数据

步骤1根据所有蛋白质的基因表达数据,结合式(1)~式(3)计算所有蛋白质的活性周期T(P),然后对计算结果进行降序排列并采用列表存储,记为:T(P)=[T1(P),T2(P),…,Tk(P)]。
步骤2根据蛋白质的活性周期构造子网:
ForTi(P),i=1,2,…,kinT(P):
在Ti(P)中计算JS(Pi,Pj);

步骤3重复执行步骤2,直到列表T(P)为空,算法结束。
2 蛋白质功能预测算法
在上文构建得到的动态蛋白质网络基础上,提出一种改进的蛋白质未知功能预测算法IPA-PF。首先对待预测功能的蛋白质在T个蛋白质子网中出现的邻居节点进行统计,然后根据其邻居蛋白质的功能已知与否,分情况进行处理。
1)如果待预测功能的蛋白质其所有邻居节点的全部功能或部分功能已知,则根据待预测功能的蛋白质与邻居蛋白质之间的连接强度来筛选参与功能预测的邻居蛋白质数目,然后通过计算候选功能得分和排序等操作实现蛋白质的未知功能预测。相关定义及具体过程如下:
定义4(功能关联得分) 设SG={G1,G2,…,GT}是基于进化图构建得到的T个蛋白质子网,Gi=(Vi,Ei,ti)。α是一个待预测的功能未知的蛋白质,β是一个功能已知的蛋白质,则β在预测α功能时的功能关联得分为:
(6)
设NS={P1,P2,…,Pn}是根据式(6)预测α的功能时形成的邻居蛋白质集合,F={f1,f2,…,fm}是NS集合中所有蛋白质的已知功能集合。设fi是F中某一蛋白质的候选功能,fi的得分为:
(7)
其中,j=1,2,…,m。对NS中所有蛋白质的候选功能根据式(7)的得分进行降序排列,并从中选取前R项功能作为蛋白质α的未知功能列表。本文算法统计NS中每一个蛋白质拥有的功能注释数量,取其中所有蛋白质的功能注释数量的最小值作为R的取值。最后,将各个邻居蛋白质的已知功能注释的交集作为待预测蛋白质α的功能。例如,对于α的邻居蛋白质{P1,P2,P3,P4}而言,蛋白质P1拥有功能{f2,f3,f7,f8},蛋白质P2拥有功能{f1,f2,f3,f6},蛋白质P3拥有功能{f2,f3,f5,f9},蛋白质P4拥有功能{f2,f3,f11,f13},因此,可以预测α拥有的功能为{f2,f3}。
2)如果待预测功能的蛋白质其所有邻居蛋白质节点的全部功能未知,则通过构建一个三层神经网络[16](包含输入层、隐藏层和输出层)模型来进行功能预测,如图3所示。

图3 基于三层神经网络的蛋白质功能预测过程Fig.3 Process of protein function prediction based onthree-layer neural network

本文提出的动态蛋白质网络蛋白质未知功能预测算法描述如下:
算法2蛋白质未知功能预测算法IPA-PF

输出未知蛋白的功能注释
步骤1对于每一个待预测功能的蛋白质α,统计其在SG中出现的邻居蛋白质节点,记为集合NS={P1,P2,…,Pk}。
步骤2如果NS中蛋白质的全部功能或部分功能已知,则:
1)根据式(6)和式(7)计算NS中所有蛋白质的候选功能得分,并对得分进行降序排列,取前R项。
2)计算NS中所有蛋白质前R项功能的交集,然后转步骤4。
步骤3如果NS中蛋白质的全部功能未知,则训练一个神经网络进行蛋白质功能预测:
1)数据预处理:采用丢弃、填充、替换或去重等操作对蛋白质的特征做归一化处理。
2)在(0,1)区间内随机初始化网络中的所有连接权值和阈值。
3)根据蛋白质的特征,采用累积误差逆传播算法[18]进行训练,得到一个连接权值与阈值确定的三层前馈神经网络(3-FNN)。
4)采用3-FNN进行蛋白质功能预测。
步骤4输出未知蛋白质的功能注释。
3 实验
实验利用Python语言实现本文提出的动态蛋白质网络构建算法和蛋白质未知功能预测算法IPA-PF。为验证动态蛋白质网络构建算法的合理性和IPA-PF的有效性,在多个数据集上将IPA-PF算法与目前较为典型的蛋白质功能预测算法D-PIN[9]、PFP-BMD[10]、PEFM[11]和HPMM[12]进行性能比较。在一台8核16线程的计算机上进行实验。其中,CPU型号为Intel Core i9-9960X@3.10 GHz,内存为16 GB,操作系统为Ubuntu 16.04 LTS 64位系统,采用GPU加速技术和TensorFlow框架来训练文中用到的神经网络,GPU型号为GeForce RTX 2070。
3.1 实验数据集
本文采用DIP数据集、MIPS数据集、GO数据库[19]和CYC数据集[20]作为测试数据集。其中,DIP数据集记录了通过生物实验测定的蛋白质之间的相互作用,它将来自各种来源的信息相互结合,形成一组单一、一致的蛋白质-蛋白质相互作用。本文使用的DIP数据是DIP20170205版本,选取其中的酵母蛋白质网络来进行实验。用UniProtKB/Swiss-Prot[21]对PPI网络中的蛋白质进行ID转换,然后去除网络中自相互作用、重复相互作用及无法转换的蛋白质后,该网络中还有4 995个蛋白质和21 554条边。MIPS数据集源自慕尼黑蛋白质序列信息中心,本文采用和上述相同的方法进行数据预处理,最终得到的相互作用网络包括4 546个酵母蛋白质和12 319对可靠的相互作用。下载基因本体(Gene Ontology,GO)数据库的最新版本来测试不同算法在蛋白质功能预测方面的性能。其中包含细胞组件、分子功能和生物过程3个独立的子本体。为保证功能预测的全面性和高效性,本文保留未被GO术语注释的蛋白质,并且保留功能注释数目不超过200个蛋白质的GO Term来进行算法验证。此外,将CYC2008作为基准数据集来评估蛋白质复合物的识别结果。该数据集中包含408个通过生物方法预测到的蛋白质复合物,每个复合物包含两个或两个以上蛋白质。
3.2 评价指标
本文采用以下指标来评价不同算法的性能:
1)查全率、查准率和F-measure值。查全率(Recall)为预测的蛋白质功能与实验数据集中真实存在的蛋白质功能注释的最大匹配数目与实验数据集中真实存在的蛋白质功能注释总数的比值,查准率(Precision)为预测的蛋白质功能与实验数据集中真实存在的蛋白质功能注释的最大匹配数目与实验测得的蛋白质功能注释总数的比值,这两个指标的计算公式如下:
(8)
(9)
其中:ER表示本文算法预测的蛋白质功能;RR表示实验数据集中真实存在的蛋白质功能注释;MNM(ER,RR)表示ER和RR之间的最大匹配数目。综合考虑查全率和查准率两方面,可得F-measure的计算公式为:
(10)
2)鲁棒性。目前能够获得的蛋白质相互作用数据都在一定程度上存在假阳性和假阴性的问题。因此,一个优秀的蛋白质构建算法和功能预测算法应对数据中存在的假阳性和假阴性具有很好的鲁棒性。
3)时间开销。在多个数据集上衡量动态蛋白质网络构建算法和蛋白质功能预测算法运行所耗费的时间,比较不同算法的运行效率。
3.3 实验结果与分析
3.3.1 IPA-PF算法与其他算法的比较
为全面分析本文提出的动态蛋白质网络构建算法和IPA-PF算法的性能,将IPA-PF算法与D-PIN[9]、PFP-BMD[10]、PEFM[11]和HPMM[12]在DIP数据集和MIPS数据集上进行比较。采用十折交叉验证法进行实验评估,即将DIP数据集和MIPS数据集分别分成10份,轮流将其中9份作为训练数据,将1份作为测试数据。为进一步降低实验误差,重复进行100次实验,取其平均值作为最终的结果。表1和表2分别列出了不同算法在DIP数据集和MIPS数据集上的性能比较。

表1 不同算法在DIP数据集上的性能比较Table 1 Performance comparison of different algorithmson DIP dataset

表2 不同算法在MIPS数据集上的性能比较Table 2 Performance comparison of different algorithmson MIPS dataset
从表1和表2的结果可以看出,本文算法在两种数据集上的查全率和查准率都要优于其他4种算法,并且在DIP数据集上,本文算法的F-measure值较HPMM、D-PIN、PEFM和PFP-BMD分别提高约40%、30%、26%和16%,在MIPS数据集上,本文算法的F-measure值较HPMM、D-PIN、PEFM和PFP-BMD分别提高约39%、26%、25%和11%,主要原因如下:
1)本文算法在构建动态蛋白质网络的过程中考虑了蛋白质基因表达的活性周期,能够更好地模拟蛋白质“合成-降解-凋亡”这一个生物过程,避免了网络构建的片面性。
2)通过引入连接强度这一概念,从物理位置上对蛋白质节点之间的相互作用进行评价,从而有效过滤了蛋白质相互作用数据中所隐含的假阳性和假阴性。
3)在未知蛋白的功能预测方面,本文对D-PIN算法的不足之处进行了改进,对待预测蛋白质节点的邻居蛋白质节点分情况(有功能注释/无功能注释)进行处理,并考虑蛋白质的多种特征来训练神经网络进行功能预测,解决了当邻居蛋白质节点的功能集合全部未知时无法进行预测这一难题,因此,本文算法能够更全面地预测蛋白质的未知功能。
3.3.2 参数th对蛋白质复合物识别性能的影响分析
在动态蛋白质网络构建过程中,参数th对于衡量两个蛋白质之间是否具有相互作用起到关键作用,下面以CYC2008数据集为实验对象,测试th取不同数值时构建出的网络在蛋白质复合物上的识别性能,选取两种典型的蛋白质复合物识别算法(MPC-TPW[22]和DPC-NADPIN[23])来分析本文构建网络算法的可靠性,实验结果如图4所示。可以看出:随着th取值增大,MPC-TP算法和DPC-NADPIN算法的F-measure值呈现不断增加的趋势,这表明两种算法能够准确识别的蛋白质复合物数量越来越多;但在th取值达到0.7之后,MPC-TP算法和DPC-NADPIN算法的性能趋于稳定,这表明本文提出的动态蛋白质网络构建算法对于输入参数不敏感,能够应用到不同的蛋白质复合物识别算法中。

图4 不同蛋白质复合物识别算法的参数敏感性比较Fig.4 Parameter sensitivity comparison of differentprotein complex recognition algorithms
3.3.3 鲁棒性分析
测试IPA-PF算法对于包含假阴性和假阳性的蛋白质相互作用数据的鲁棒性。以DIP数据集为测试用例,在实验中通过随机增加和删除一定比例的边来模拟蛋白质网络的假阳性和假阴性。其中:假阳性是指能够被实验技术检测到但在细胞中并不存在的蛋白质相互作用;假阴性是指不能被实验技术检测到但在细胞中确实存在的蛋白质相互作用。以每20个百分点为一个间隔,随机地增加边的比例从20%到100%,共得到5组数据,从这些具有较高假阳性的数据中识别蛋白质复合物,得到IPA-PF算法的查全率和查准率,如图5所示。可以看出,随着假阳性的增强,IPA-PF算法预测蛋白质功能的查全率基本保持不变,而查准率有轻微下降,这表明IPA-PF算法具有较强的抗噪能力,能够应对那些被算法检测得到但在数据集中并不存在的蛋白质相互作用。

图5 数据包含假阳性时IPA-PF算法的性能指标Fig.5 Performance indexes of IPA-PF algorithmwith false positive data
以每20个百分点为一个间隔,随机地删除边的比例从15%到90%,共得到6组数据,重复上述工作,得到IPA-PF算法的查全率和查准率,如图6所示。可以看出:当删除边的比例小于45%时,IPA-PF算法预测蛋白质功能的查全率和查准率基本保持不变;在删除边的比例超过40%后,IPA-PF算法的性能开始呈现直线下降趋势,这是因为随着假阴性的增强,数据集中那些未被IPA-PF算法检测到但又真实存在的相互作用会被大量删除,理论上会使算法能够预测的蛋白质功能数量急剧减少,而IPA-PF算法反映在查全率和查准率上的变化就是这两种指标直接降低,这也恰好验证了IPA-PF算法对于假阴性具有较好的鲁棒性。

图6 数据包含假阴性时IPA-PF算法的性能指标Fig.6 Performance indexes of IPA-PF algorithm withfalse negative data
3.3.4 不同算法的效率分析
为进一步衡量本文算法的优越性,在上述实验环境下对不同蛋白质功能预测算法的时间开销进行测试。以DIP数据集和MIPS数据集作为测试用例,表3给出了不同算法在进行蛋白质未知功能预测时的运行时间。可以看出,IPA-PF算法在两种数据集上的运行时间均不超过11 s,低于D-PIN、PEFM和HPMM算法,略高于PFP-BMD算法。但通过上文的实验分析结果可知,IPA-PF算法的预测质量远超其他预测算法。从性能折中的角度来看,以目前计算机的算力而言,在保证蛋白质功能预测准确性的前提下,牺牲算法的部分效率完全是可以接受的。总体而言,本文提出的IPA-PF算法具有较高的运行效率,可适用于大规模的蛋白质网络。

表3 不同算法的运行时间比较Table 3 Running time comparison ofdifferent algorithms s
4 结束语
蛋白质相互作用网络是目前蛋白组学的研究热点。针对现有蛋白质网络构建和功能预测方法存在的不足,本文提出一种基于进化图的动态蛋白质网络构建算法,在此基础上设计一种新的蛋白质功能预测算法,并在多个公开的生物数据库上验证算法的有效性。本文研究有利于从微观层面解释细胞内蛋白质之间的复杂关系,为生物学和医学领域研究者理解生命复杂网络的内在组织和生物过程提供了新的途径,并可用于药物标靶设计、疾病诊治和预测等多个方面。下一步将分析影响动态蛋白质网络构建的诸多因素,并采用深度学习技术对关键蛋白质的识别进行建模,设计基于图卷积神经网络的关键蛋白质识别算法。
猜你喜欢
黄河之声(2022年10期)2022-09-27
卫星应用(2022年7期)2022-09-05
肝博士(2022年3期)2022-06-30
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25
中学生数理化(高中版.高二数学)(2022年4期)2022-05-25
卫星应用(2022年3期)2022-05-23
卫星应用(2022年1期)2022-03-09
海外星云(2021年9期)2021-10-14
环球慈善(2019年6期)2019-09-25
中学生数理化·八年级物理人教版(2017年11期)2017-04-18
