
洋甘菊多糖的分离纯化、性质结构及抗氧化活性分析
2021-02-25 13:59陆娟谢东雪贺柳洋王月郑志艳
食品与发酵工业 2021年3期
陆娟,谢东雪,贺柳洋,王月,郑志艳
(长春师范大学 化学学院,吉林 长春,130032)
洋甘菊(MatricariachamomillaL.),一年生或两年生草本植物,又称为母菊,原产于欧洲各国,栽种于德国、法国和摩洛哥等地,目前在欧洲、亚洲等地比较常见[1],是一种重要的药用植物和香料植物[2],在世界范围内广泛栽培和消费,具有消炎、抑制真菌等作用[3]。目前,已经从洋甘菊中分离得到具有抗炎、抗菌、抗过敏[4]活性的挥发油、黄酮、香豆素等化合物。
文献中关于洋甘菊化学成分的报道大都集中在小分子化合物,而关于洋甘菊中多糖的报道却比较少。课题组在2019年对洋甘菊多糖的提取工艺进行了优化,并研究了粗多糖的抗氧化活性[5]。本文在之前研究的基础上,继续利用传统的热回流法提取洋甘菊多糖,通过凝胶过滤色谱分离纯化洋甘菊多糖,得到2种均一多糖组分,并研究其理化性质及抗氧化活性,为洋甘菊多糖进一步研究及开发利用提供依据。
1 材料与方法
1.1 材料与试剂
洋甘菊购于新疆维吾尔自治区;DEAE琼脂糖凝胶CL-6B、葡聚糖凝胶G-100,美国GE Healthcare;Dextran对照品、系列单糖标准品、1,1-二苯基-2-三硝基苯肼(1,1-diphenyl-2-picrylhydrazyl,DPPH)、1-苯基-3-甲基-5-吡啉唑酮(1-phenyl-3-methyl-5-pyrazolone,PMP),上海阿拉丁生化科技股份有限公司。液相色谱用乙腈、甲醇均为色谱纯,其他试剂均为分析纯。
1.2 主要设备
UV—1901紫外可见分光光度计,北京普析仪器公司;中压快速纯化制备系统,科穗科技(苏州)有限公司;Waters 2695高效液相色谱仪(配备2998 PDA、2414示差折光检测器),美国沃特世公司;Nicolet iS50 型红外光谱仪,赛默飞世尔科技公司;TG-7热重分析仪,美国Pekin-Elmer公司。
1.3 实验方法
1.3.1 洋甘菊多糖的制备
干燥粉碎的洋甘菊粉末先后用石油醚、体积分数95%乙醇回流2 h提取小分子化合物,晾干后,密封备用。称取经上述处理的50 g粉末,按照液料比30∶1(mL∶g)加入1 500 mL蒸馏水浸泡12 h,分多次回流提取1次,每次2 h。回流液经离心(5 000 r/min,10 min)后减压浓缩一定体积,加入适量的无水乙醇使样品中乙醇体积分数约为85%,将上述醇沉液于4 ℃冰箱中静置12 h后离心(5 000 r/min,10 min),保留沉淀;沉淀加入适量蒸馏水重新溶解, Sevag法除蛋白,H2O2法脱色,透析袋流水透析除杂,最后浓缩冻干得到的洋甘菊粗多糖粉末,命名为MCP。
1.3.2 洋甘菊多糖的分离纯化
准确称取MCP粉末200 mg,加入适量超纯水,使样品尽可能地溶解,且样品溶液不宜过多,利用0.45 μm水系滤头过滤样品溶液后,直接滴加到已经处理好的DEAE-Sepharose色谱柱(30 mm×100 mm),待样品滴加完毕后将色谱柱连接到中压快速纯化制备系统,分别用0、0.5和1.0 mol/L NaCl溶液洗脱,自动收集洗脱液(10 mL/管,2 mL/min)。在整个洗脱过程中利用苯酚-硫酸法[6]测定收集液的吸光度值,绘制吸光度值与洗脱管数的关系曲线,根据洗脱曲线收集单一对称峰,冷冻干燥。为得到足够量的样品反复进行上述操作5次。
分别称取经DEAE-Sepharose色谱柱分离得到的样品200 mg,加入适量超纯水溶解,经Sephadex G-100色谱柱(20 mm×150 mm)分离,超纯水洗脱,收集洗脱液(5 mL/管,1 mL/min),苯酚-硫酸法[6]检测绘制洗脱曲线等操作后,得到均一多糖。
1.3.3 均一多糖理化性质
分别采用苯酚-硫酸法[6]、硫酸-咔唑法[7]、考马斯亮蓝法[8]测定均一多糖的总糖、糖醛酸及蛋白质的含量。溶解度的测定在文献[9]的基础上稍做改动。准确称取分离得到的洋甘菊均一多糖样品0.01 g(m0)放置于离心管中,加入1 mL蒸馏水溶解,在90 ℃水浴中加热90 min,其中每30 min振荡5 s,然后高速离心10 min(10 000 r/min),最后除去上清液,将沉淀置于105 ℃下加热,直到样品质量恒定(m1)。溶解度按公式(1)计算:
(1)
1.3.4 均一多糖分子质量测定
根据课题组的研究方法[10],利用高效凝胶渗透色谱法(high performance gel permeation chromatography,HPGPC)测定均一多糖分子质量,色谱条件为:高效液相色谱仪(Waters 2695型)配备示差折光检测器(Waters 2414),3根GPC色谱柱串联,柱温35 ℃, 0.1 mol/L的NaNO3以1.0 mL/min的流速进行洗脱,进样量20 μL。分别取20 μL 10 mg/mL系列Dextran 对照品溶液(分子质量分别为5、12、25、50、80、270、410、670 kDa)注入高效液相色谱仪,绘制Dextran 对照品分子量对数值与保留时间(tR)线性回归曲线。分别将10 mg/mL的样品溶液在同样色谱条件下进样,根据保留时间和线性回归曲线计算均一多糖分子质量。
1.3.5 均一多糖单糖组成
均一多糖的单糖组成按照课题组前期研究方法进行[11]。准确取10 mg单糖样品和2 mL 2 mol/L 的H2SO4溶液一起放入反应釜中,样品溶解、充氮气、密封反应釜,放入95 ℃恒温的烘箱中水解8 h。关闭烘箱冷却至室温,取出反应釜,倒出清澈透明上清液,加入适量6 mol/L NaOH溶液调节至中性后,加超纯水定容至2 mL。分别移取500 μL样品溶液,200 μL 0.5 mol/L NaOH溶液和200 μL 0.5 mol/L PMP甲醇溶液同置于小试管中,充氮气密封,70 ℃水浴30 min。取出冷却至室温后,再以0.5 mol/L 盐酸调节样品溶液至中性后,利用氯仿萃取未反应完的PMP,每次1 mL,萃取3~5次,上层水溶液0.45 μm微孔滤膜过滤后进行HPLC分析。取10 μL样品和标准品分别注入到配备2998 PDA检测器的高效液相色谱仪(Waters 2695),色谱柱是Venusil MP C18(2)(6 mm×250 mm),柱温35 ℃,V(乙腈)∶V(pH 6.86磷酸缓冲溶液)=17∶83作为流动相,流速0.8 mL/min,检测波长254 nm,样品和标准品均运行45 min。
1.3.6 刚果红试验
参照文献[12],分别取1 mL多糖样品(1 mg/mL)放置于7个比色管中,分别加入1 mL (80 μmol/L)刚果红溶液,再加入不同体积的1 mol/L NaOH溶液,使混合体系中的NaOH最终浓度分别为0、0.1、0.2、0.3、0.4、0.5 和 0.6 mol/L。混合体系在室温下反应25 min后,分别在400~600 nm波长范围内绘制其吸收曲线,确定体系最大吸收波长λmax,而后分别以各混合体系的NaOH终浓度为横坐标,λmax为纵坐标绘制曲线。
1.3.7 均一多糖红外光谱测定
称取适量均一多糖样品,加入一定量的KBr粉末,置于玛瑙研钵中研磨均匀,压片均匀后于4 000~400 cm-1波数范围内测定红外光谱。
1.3.8 均一多糖热稳定性分析
参照文献[13],使用热重分析仪对均一多糖进行热稳定性分析。将粉状多糖在氮气环境下以10 ℃/min的升温速率从25 ℃加热到600 ℃,绘制热重曲线。
1.3.9 洋甘菊多糖抗氧化活性
洋甘菊多糖清除DPPH自由基能力按照课题组前期研究方法[11]进行,分别移取2 mL不同质量浓度(1~6 mg/mL)均一多糖样品溶液,和2 mL DPPH(2 mmol/L甲醇溶液)混合均匀,暗处反应30 min后,在波长517 nm处测定混合溶液的吸光度Ai,同时测定DPPH溶液的吸光度ADPPH,利用 VC做为阳性对照,多糖对DPPH的清除率用SA(%)表示,按公式(2)计算:
(2)
按照课题组前期研究方法[5]进行·OH清除试验,首先取1.0 mL pH 7.4的磷酸缓冲溶液,0.2 mL 520 μg/mL番红,1.0 mL乙二胺四乙酸钠铁(Ⅱ)(EDTA Na-Fe(II))溶液混合,再加入1.0 mL(1~6 mg/mL)均一多糖样品溶液,最后加入0.8 mL体积分数为6% H2O2溶液启动反应,混匀后于37 ℃的水浴中加热30 min,于波长515 nm处测吸光度A样品,以等量超纯水替代样品作为空白组,测吸光度A空白,以等量超纯水替代样品溶液和H2O2溶液作为对照组,测吸光度A对照,多糖对·OH的清除率用SA(%)表示,按公式(3)计算:
(3)
2 结果与分析
2.1 洋甘菊多糖分离纯化
洋甘菊多糖经DEAE Sepharose CL-6B柱色谱分离,利用0.0、0.5、1.0 mol/L NaCl溶液洗脱后,洗脱曲线见图1。MCP经上述溶液洗脱后得到2个明显的洗脱峰,收集两组分,经透析、浓缩、冷冻干燥等系列操作后得到两部分样品粉末,分别命名为MCP-1和MCP-2。
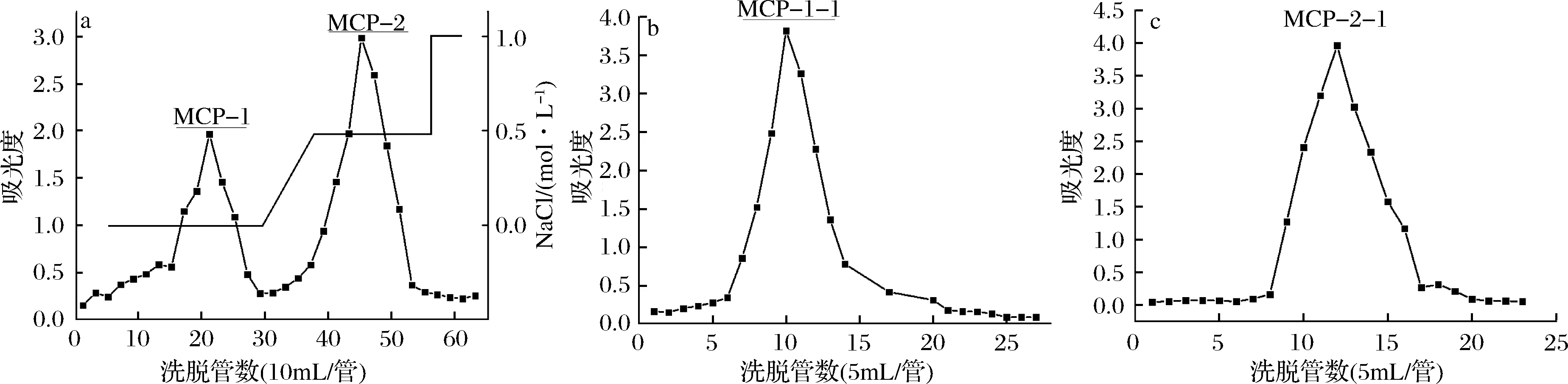
图1 洋甘菊多糖的DEAE Sepharose(a)和Sephadex G-100柱色谱洗脱曲线(b, c)Fig.1 Elution curves of MCP purified with DEAE Sepharose column (a) and Sephadex G-100 (b, c)
取MCP-1和MCP-2,继续利用Sephadex G-100色谱柱纯化,洗脱曲线见图2。MCP-1和MCP-2继续纯化后均得到单一对称峰,分别命名为MCP-1-1和MCP-2-1,收集样品,浓缩、冻干。
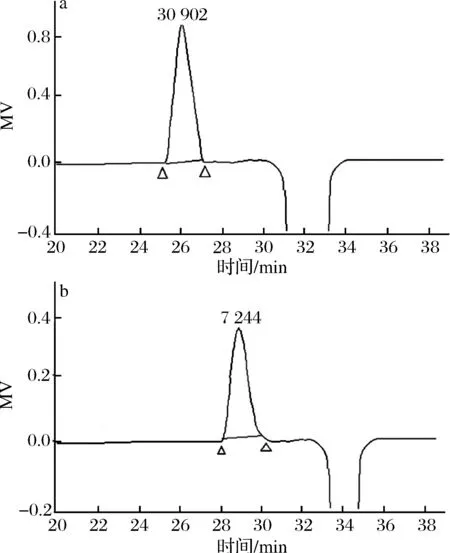
图2 MCP-1-1(a)、MCP-2-1(b)高效凝胶渗透色谱图Fig.2 HPGPC chromatogram of MCP-1-1(a) and MCP-2-1(b)
2.3 多糖分子质量测定结果
MCP-1-2和MCP-2-1的HPGPC图见图2。2个HPGPC图均是单个对称峰,而且峰形相对对称,可以认为MCP-1-1和MCP-2-1为均一多糖。通过HPGPC法测定均一多糖的分子质量,以葡聚糖标准建立的标准曲线为:lgMw=-0.232 4tR+9.964 9 (R2=0.987 8),Mw是多糖的分子质量,而tR是保留时间。均一多糖MCP-1-1和MCP-2-1的tR分别为26.38和29.38 min,计算其分子质量分别为30 902和7 244 Da。
2.4 单糖组成
图3为混合标准单糖和MCP-1-1、MCP-2-1经水解、PMP衍生后的HPLC图。由图3可知,MCP-1-1是杂多糖,是由Rha、GlcA、Gal、Glc、Xyl和Ara六种单糖组成,经计算其摩尔百分比(%)分别为7.29、2.74、4.31、14.89、12.73、55.23。MCP-2-1由GlcA、Gal、Glc、GalA、Xyl和Ara六种单糖组成,其摩尔百分比(%)分别为5.53、52.88、4.26、12.24、10.21、6.32。
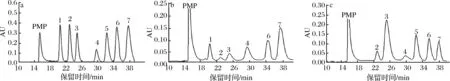
1-Rha, 2-GlcA,3-Gal, 4-Glc, 5-GalA, 6-Xyl, 7-Ara图3 混合标准单糖(a)和MCP-1-1(b)、MCP-2-1(c)酸水解后HPLC图Fig.3 The HPLC chromatograms of 7 standard monosaccharides MCP-1-1 and MCP-2-1
2.5 刚果红实验结果分析
刚果红可以与具有三重螺旋结构多糖络合,生成络合物的最大吸收波长与刚果红溶液的最大吸收波长相比将会发生红移[14],而且随着NaOH浓度的增加,其最大吸收波长先增大后减小[15]。图4显示MCP-1-1和MCP-2-1与刚果红混合后,混合溶液的最大吸收波长(λmax)随着NaOH最终浓度变化的曲线图。与刚果红溶液相比, MCP-1-1和MCP-2-1分别与刚果红混合溶液最大吸收波长几乎没有改变,这表明这两部分多糖溶液中不存在三重螺旋结构。有文献报道称单糖种类较多的多糖不容易形成三重螺旋结构[16],结合MCP-1-1和MCP-2-1单糖组成结果发现,两种均一多糖都是由6种不同的单糖组成,因而不具有三螺旋结构。
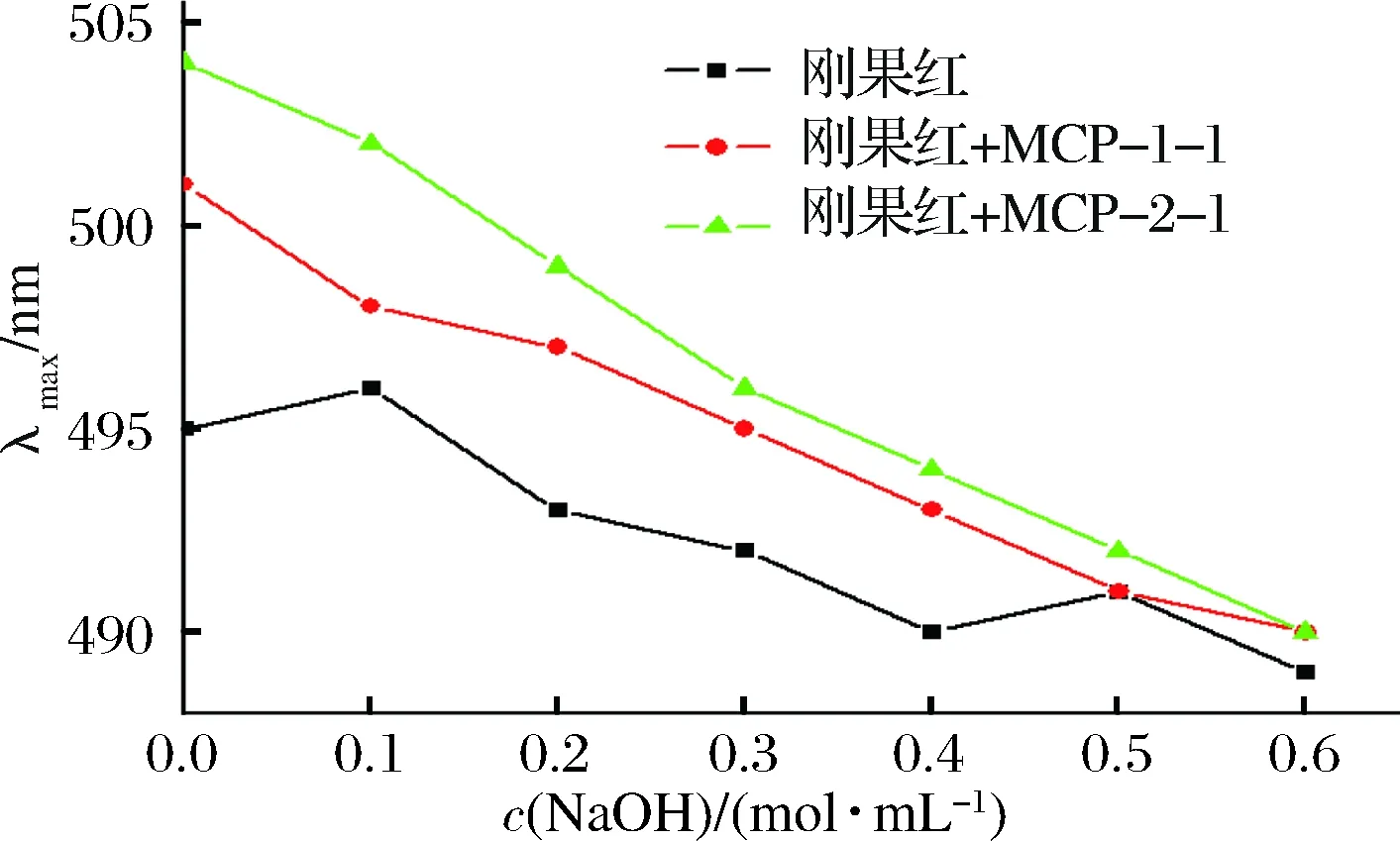
图4 MCP-1-1和MCP-2-1刚果红试验结果图Fig.4 Congo red test of MCP-1-1 and MCP-2-1
2.6 红外光谱分析结果

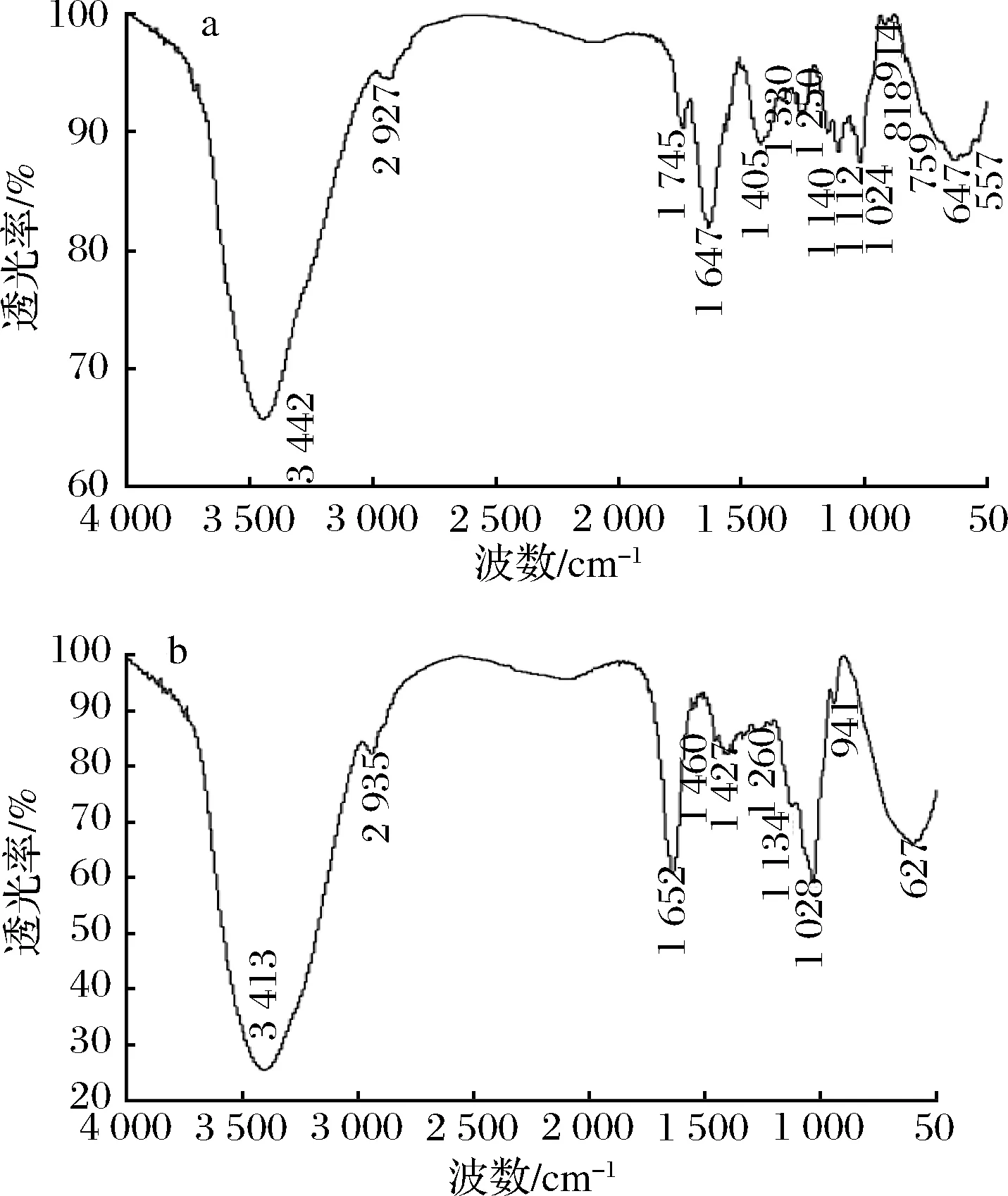
图5 MCP-1-1(a)、MCP-2-1(b)红外光谱图Fig.5 IR spectra of MCP-1-1(a) and MCP-2-1(b)
2.7 热稳定性结果分析
如图6所示,两部分多糖组分的热重曲线分为3个不同的阶段。MCP-1-1(a)和MCP-2-1(b)第1阶段失重范围分别为25~123、32~130 ℃,主要是由于自由水和束缚水的损失[23],质量损失率约为8.7%和6.3%,结果表明MCP-1-1比MCP-2-1有更高的保水能力。第2次快速失重阶段分别出现在123~375和130~394 ℃, MCP-1-1和MCP-2-1的失重分别为24.6%和17.8%,这可能与多糖的热分解有关[24]。第3次失重范围为375~791和394~793 ℃时,由于碳的热分解作用,使其质量逐渐下降[25]。结果表明,MCP-2-1较MCP-1-1表现出良好的热稳定性,可用于高温加工的食品和药品中。
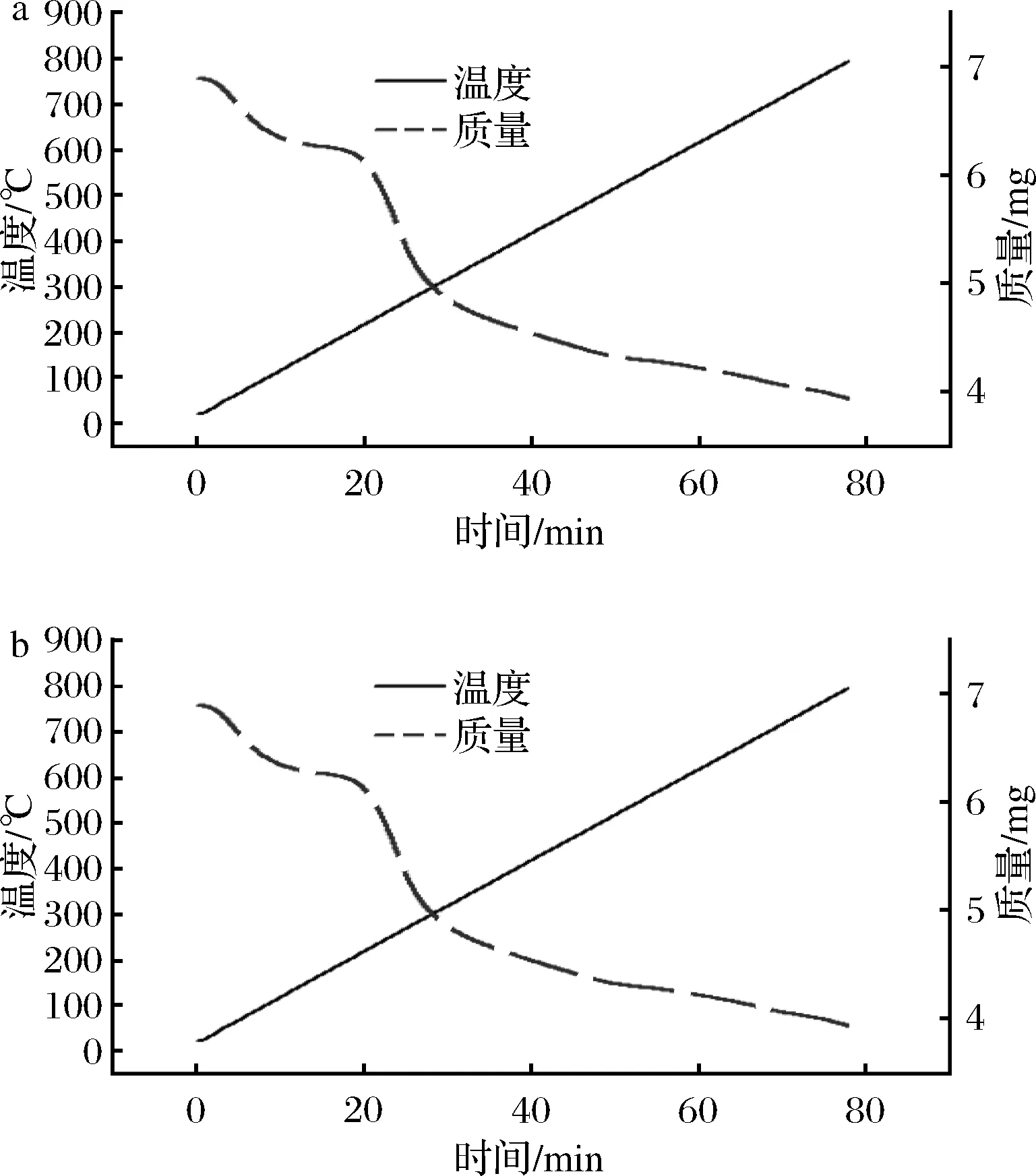
图6 MCP-1-1(a)和MCP-2-1(b) 热量曲线Fig.6 TG curve of MCP-1-1(a) and MCP-2-1(b)
2.8 洋甘菊多糖体外抗氧化活性结果
MCP-1-1、MCP-2-1及Vc对DPPH自由基的清除作用结果如图7-a所示,在1~6 mg/mL时,多糖样品对DPPH自由基的清除能力随着质量浓度的增加而变大,但一直低于Vc的清除效果。MCP-1-1和MCP-2-1清除DPPH自由基的半数抑制浓度(IC50)值分别为:1.67(MCP-2-1)、2.95(MCP-1-1)mg/mL。这可能是由于MCP-2-1分子质量仅为7 244 Da,要小于MCP-1-1,此外,MCP-2-1中糖醛酸含量也高于MCP-1-1,由此可见,多糖分子质量和糖醛酸含量均可能是影响多糖生物性的主要因素,分子质量越小生物活性越高,糖醛酸含量越高,生物活性越高。
如图7-b所示,质量浓度在1~6 mg/mL时,MCP-1-1和MCP-2-1对·OH的清除率与质量浓度呈现正相关,当质量浓度为6 mg/mL时,MCP-1-1和MCP-2-1的清除率分别为53.4%和84.9%。MCP-1-1、MCP-2-1的IC50值分别为5.33和3.21 mg/mL。MCP-2-1对·OH的清除率明显高于MCP-1-1,但低于Vc的清除能力。这也验证了多糖分子质量、糖醛酸含量可能是影响多糖的生物活性的主要因素。
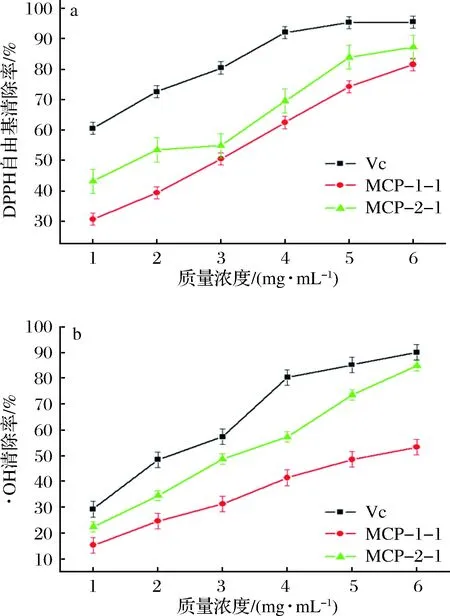
a-DPPH自由基;b-·OH图7 MCP-1-1和MCP-2-1清除自由基能力Fig.7 Free radical scavenging ability of MCP-1-1 and MCP-2-1
3 结论
热回流提取的洋甘菊粗多糖经DEAE-Sepharose CL-6B柱色谱和Sephadex G-100柱色谱分离纯化,得到MCP-1-1和MCP-2-1两个均一多糖组分。经测定MCP-1-1和MCP-2-1总糖含量为 82.23%、84.09%,糖醛酸含量为4.11%、5.38%,蛋白质含量为0.58%、1.05%,溶解度为98.36%、97.44%,分子质量分别为30 902、7 244 Da。单糖组成分析表明 MCP-1-1是由Rha、GlcA、Gal、Glc、Xyl和Ara六种单糖以摩尔百分比(%)分别为7.29、2.74、4.31、14.89、12.73和55.23组成的。MCP-2-1是由GlcA、Gal、Glc、GalA、Xyl和Ara六种单糖以摩尔百分比(%)分别为5.53、52.88、4.26、12.24、10.21和6.32组成的,2种均一多糖均不具有三螺旋结构。红外光谱显示MCP-1-1、MCP-2-1均属于酸性多糖,含有吡喃环和β-糖苷键,其中MCP-1-1含有α-糖苷键,MCP-2-1不含α-糖苷键。热稳定结果表明温度低于120 ℃时,MCP-1-1比MCP-2-1有更高的保水能力,而高于120 ℃以后,MCP-2-1热稳定性高。体外清除自由基试验表明MCP-1-1和MCP-2-1对DPPH自由基和·OH均有清除作用,而且清除能力与均一多糖的质量浓度正相关,MCP-2-1对自由基的清除能力明显高于MCP-1-1,这可能是因其分子质量相对较低,糖醛酸含量较高引起的。本文的研究结果表明,分离纯化得到的洋甘菊多糖MCP-1-1、MCP-2-1均可作为天然抗氧化剂用于健康食品和化妆品加工中,而MCP-2-1还可用于高温加工的食品和药品中,研究结果为洋甘菊进一步的开发利用提供一定的依据。
猜你喜欢
中老年保健(2021年9期)2021-08-24
农村青少年科学探究(2020年5期)2020-08-18
中成药(2019年12期)2020-01-04
天然产物研究与开发(2019年1期)2019-03-01
中央民族大学学报(自然科学版)(2018年1期)2018-06-27
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
中成药(2017年7期)2017-11-22
少儿科学周刊·少年版(2015年1期)2015-07-07
食品工业科技(2014年13期)2014-03-11
