
NO供体型大黄酸衍生物的合成及抗肿瘤活性
2021-03-13 06:53柏志伟尚飞扬戴卫国何黎琴
中国药科大学学报 2021年1期
柏志伟,尚飞扬,戴卫国,何黎琴
(安徽中医药大学药学院,合肥230031)
大黄在我国具有悠久的药用历史,大黄酸(rhein,RH)是其有效活性成分之一。研究发现,大黄酸具有广泛的药理活性,尤其在抗炎、抗肿瘤等方面表现突出[1-3]。研究表明,大黄酸可用于多种肿瘤性疾病的治疗,具有广谱的抗肿瘤作用,且作用机制多样:大黄酸的蒽醌三环共平面结构可以嵌入到DNA 的碱基对中,与DNA 双螺旋结构可逆性的结合,从而导致DNA 裂解和影响DNA 的转录、合成,达到抗肿瘤的作用;能抑制大鼠肝细胞的能量代谢,影响肝细胞的呼吸链,造成肝细胞内过氧化物积累,最后造成肝细胞死亡;抑制人表皮生长因子的双靶点受体蛋白酪氨酸激酶的活性,达到抗乳腺癌的目的;能与细胞内的转录激活因子(AP-1)结合,通过AP-1 来调节结肠细胞对细胞毒剂的敏感性,达到抗肿瘤的作用;通过下调多药耐药相关蛋白1 的表达,逆转肿瘤细胞多药耐药性[4-8]。此外,大黄酸具有毒性低、安全性高等优势,为研发高效、低毒、多靶点的大黄酸类抗肿瘤药物提供了良好的先导化合物。由于大黄酸抗肿瘤活性不够强,溶解性能差,既不溶于水和醇,又不溶于绝大多数有机溶剂,生物利用度较低,使大黄酸的临床使用受到了很大限制。鉴于大黄酸的众多药理作用和明显的缺点,加之结构简单、可修饰位点较多,通过对其结构进行修饰和改造来改善大黄酸的溶解性能,增加其水溶性或脂溶性,提高其生物利用度和抗肿瘤活性,已成为当前的研究热点。多个课题组对其进行了结构修饰,取得了很好的效果,得到的系列大黄酸衍生物在抗肿瘤活性、溶解性能和生物利用度方面均具有较大的改善,为其进一步研发打下良好的基础[9-14]。
NO 作为重要的信使物质或效应分子,参与体内多种生理和病理反应。大量研究表明,体内高浓度的NO 可产生细胞毒性,诱导肿瘤细胞凋亡,阻止肿瘤细胞的扩散和转移,促进巨噬细胞杀死肿瘤细胞。研究发现,NO 供体型衍生物在药效或安全性方面较先导化合物有明显的改善[15-17]。受此启发,本课题组前期设计合成了系列NO 供体型苦参碱和香豆素的衍生物[18-21],体外抗肿瘤活性强于母体化合物,部分化合物活性优于阳性对照药氟尿嘧啶。在此基础上,利用药物化学的拼合原理,本课题组通过大黄酸的2 位羧基与硝酸酯类NO供体偶联,设计合成了5个硝酸酯类NO供体型大黄酸衍生物,其抗肿瘤活性显著高于先导化合物大黄酸,部分化合物的抗细胞增殖作用强于氟尿嘧啶[22]。受此鼓舞,为获取更多的具有抗肿瘤活性的大黄酸衍生物,本研究拟选用另一类重要的NO 供体——呋咱氮氧化合物,通过不同的连接臂,将其与大黄酸-2-位羧基偶联,设计合成了7 个目标化合物。期望得到的化合物能改善大黄酸的溶解性,提高生物利用度,并能够在体内通过协同效应增强其抗肿瘤活性。
1 合成路线
以大黄酸(1)为原料,经二氯亚砜卤代得到大黄酰氯(2),中间体2 再与不同的羟烃氧基取代的呋咱(3a~3g)反应得到目标化合物(4a~4g)。合成路线见路线1。
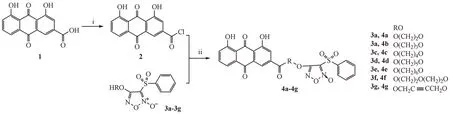
Scheme 1 Synthetic routes of compounds 4a-4g
2 实验部分
2.1 仪器与试剂
LCQ ADVANTAGE MAX 液质联用质谱仪(美国Finnigan 公司);Nicolet Acatar 370 DTGS 型红外光谱仪(美国Thermo Electron 公司);AV 400 型核磁共振仪(德国Bruker 公司,溶剂为氘代二甲基亚砜);薄层硅胶G板(100 mm×100 mm,合肥森瑞有限公司);大黄酸(含量大于98%,西安小草植物科技有限责任公司;一氧化氮检测试剂盒(上海碧云天生物技术有限公司);血红蛋白(上海碧云天生物技术有限公司)。其他试剂均为市售分析纯。
2.2 化学合成
2.2.1 中间体(2)的合成 在干燥的100 mL圆底烧瓶中,加入大黄酸(1 g,3.5 mmol)和二氯亚砜(30 mL),DMF0.5 mL,70 ℃回流搅拌6 h,减压蒸去氯化亚砜,得橘红色固体1.1 g(不经纯化,直接用于下步反应)。
2.2.2 中间体3a~3g 的合成 参照文献[21]将苯硫酚(12.1 g,0.11 mol),氢氧化钠(4.4 g,0.11 mol)溶于95%乙醇50 mL 中,加入由氯乙酸(11.4 g,0.12 mol)和碳酸钠(6.35 g,0.06 mol)配成的水溶液100 mL,室温搅拌3 h,回流1 h。冷却至室温后加入6 mol/L 盐酸调至pH 2,减压蒸去乙醇,有白色沉淀生成,过滤,得白色棒状晶体2-苯硫基乙酸16.4 g,收率89%,mp:60.1~62.0 ℃。
将2-苯硫基乙酸(16.0 g,0.1 mol)溶于冰醋酸65 mL中,加入30%过氧化氢(20 mL,0.2 mol),室温搅拌2.5 h,得无色澄清溶液,滴加95%发烟硝酸(40 mL,0.9 mol)升温至90 ℃反应30 min,冷却至室温,有白色针状晶体3,4-二苯磺酰基-1,2,5-■二唑-2-氧化物析出,过滤干燥得14 g,两步收率76%,mp:154.2~156.0 ℃。
将相应的二醇(10 mmol)和3,4-二苯磺酰基-1,2,5-■二唑-2-氧化物(1 g,2.7 mmol)溶于THF 10 mL 中,滴入25%氢氧化钠溶液(0.5 mL,3 mmol),2 h 后,反应液从淡黄色变为橙黄色。将反应液倾入水20 mL中,用乙酸乙酯(20 mL×3)萃取,有机层合并后加饱和氯化钠溶液水洗一次,用无水硫酸钠干燥。过滤后将滤液浓缩,柱色谱[乙酸乙酯-石油醚(60~90 ℃),1∶4],得白色粉末状固体3a~3g。
2.2.3 目标化合物4a~4g 的合成 将中间体3(1.0 mmol)、吡啶(0.24 mL,3.0 mmol)溶于二氯甲烷(DCM)20 mL 中,在冰盐浴中缓慢滴加大黄酸酰氯(453 mg,1.5 mmol)的二氯甲烷溶液,滴加完毕,升至室温搅拌,TLC检测反应进程。反应完毕,加水100 mL,DCM 萃取(20 mL × 3),有机相合并后用饱和氯化钠溶液洗涤1 次,无水硫酸钠干燥。过滤,滤液浓缩,柱色谱(流动相为DCM)得黄色固体化合物4a~4g。
合成的7 个目标化合物的理化常数和波谱数据分别见表1和表2。
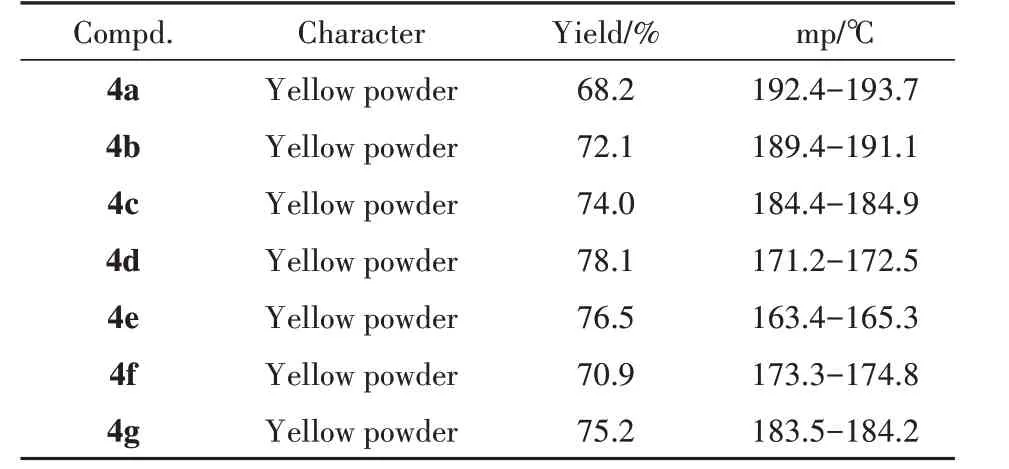
Table 1 Yield and physical properties of compounds 4a-4g

Table 2 IR, 1H NMR and MS datas of compounds 4a-4g

(Continued)
2.3 细胞毒活性测试
以先导化合物大黄酸和临床常用抗肿瘤药物氟尿嘧啶(5-FU)为阳性对照,采用MTT 法测试目标化合物对人肝癌细胞HepG2、Bel-7402,人结肠癌细胞HCT116,人骨肉瘤细胞U2OS,耐药细胞Bel-7402/5-FU 及正常肝细胞LO2 的体外抗细胞增殖活性。细胞在37 ℃、5%CO2饱和湿度的培养箱中常规培养。取处于对数生长期状态良好的细胞,加入消化液(0.125%胰蛋白酶+0.01%EDTA)消化,制成每毫升2×104~4×104个细胞的细胞悬液,接种于96 孔板上,每孔加180 μL,置恒温CO2培养箱中培养24 h。更换培养液,加入受试药物(0.16~25 μmol/L,5 个浓度),每孔20 μL,培养48 h。将MTT加入96孔板中,每个孔20 μL,培养箱中孵育4 h。吸去上清液,加DMSO,每孔150 μL,平板摇床上振摇10 min,用酶联免疫检测仪测定每孔在波长为570 nm处的吸收度,计算IC50。
2.4 NO释放量的测定
将1×106个HepG2 细胞置于6 孔板中孵育过夜。向每孔中分别加入100 μmol/L 化合物4g 孵育150 min。收集细胞并将其裂解,在96 孔板中加入裂解液50 μL,再分别加入一氧化氮检测试剂盒中Griess 试剂Ⅰ和Ⅱ50 μL,于37 ℃孵育10 min,测定在540 nm 处的吸收度。只用DMSO 给药处理的细胞作为亚硝酸盐产物的背景的阴性对照,以不同浓度的亚硝酸钠作标准曲线,根据标准曲线计算NO的浓度。
2.5 NO清除剂对化合物4g活性的影响
用NO 清除剂血红蛋白检验NO 释放对化合物4g 活性的影响。方法参照“2.3”项,取人肝癌细胞HepG2 细胞,将血红蛋白(终浓度10 μmol/L,未见显著的细胞毒性)先于受试药物1 h 加入培养基,再用受试药物4g(终浓度12.5 μmol/L)处理72 h,采用MTT法检测细胞抑制率。
3 结果与讨论
3.1 合成部分
在合成目标化合物的过程中,本研究曾参考前期类似化合物的合成方法,即以大黄酸和羟烃氧基取代的呋咱在脱水剂EDCI/DMAP 作用下一步反应成酯,实验发现产率低,且后处理操作繁琐。为此,改用酰氯成酯的方法,即大黄酸经二氯亚砜卤代得到大黄酰氯(2),再与不同的羟烃氧基取代的呋咱反应得到目标化合物(4a~4g)。考虑到所制备的大黄酰氯没有经过精制处理,在设计反应物配比时,加大了酰氯的用量。多次的实验表明,大黄酰氯与羟烃氧基呋咱物质的量比1.5∶1为最佳,反应温度为先冰盐浴中滴加酰氯,后室温反应。实验结果表明,以此法制备目标化合物,反应能较好地进行,且后处理方便,反应收率较高,可达68%以上。
3.2 细胞毒活性
为了研究目标化合物的抗肿瘤活性,以先导化合物大黄酸和抗肿瘤药物5-FU 为阳性对照,采用MTT 法测试了目标化合物对人肝癌细胞HepG2,Bel-7402,人结肠癌细胞HCT116,人骨肉瘤细胞U2OS,耐药细胞Bel-7402/5-FU 及正常肝细胞LO2的体外抗细胞增殖活性。实验结果见表3。
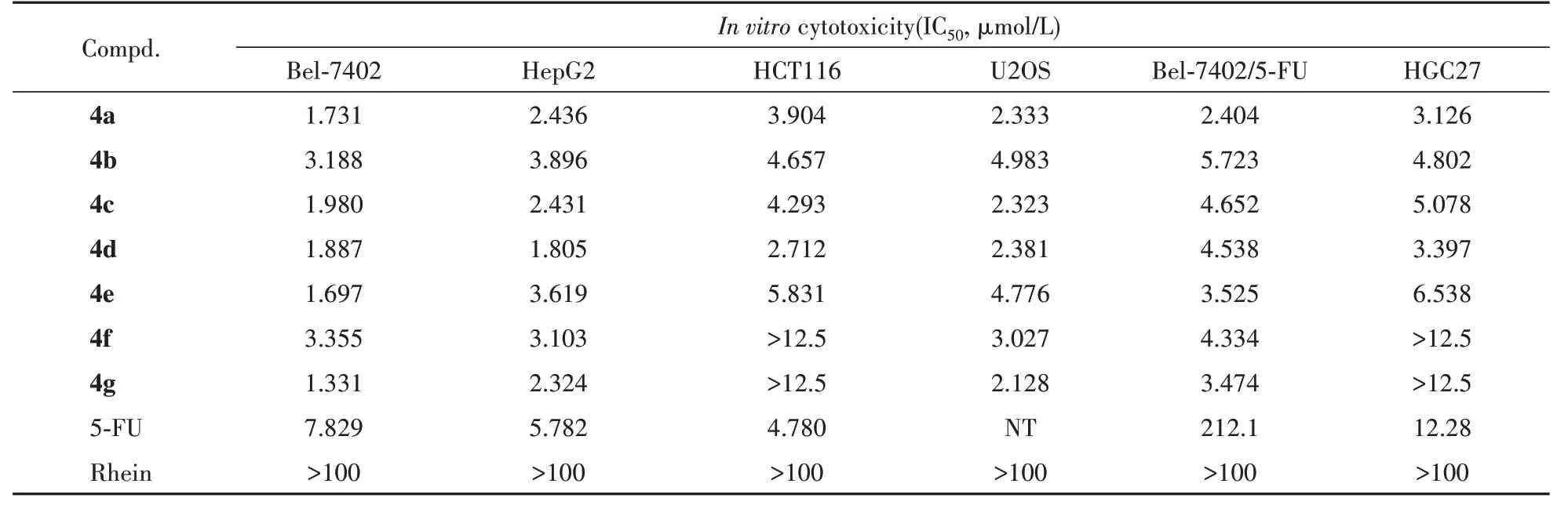
Table 3 Effects of target compounds against six human cancer cell lines
由表3 可知,大黄酸呋咱偶联物对所测试的6 种人类肿瘤细胞均表现出一定的增殖抑制活性,且全部目标化合物对肿瘤细胞的抑制活性远高于先导化合物大黄酸,绝大部分目标化合物的抗肿瘤活性与阳性对照药5-FU相当或更强。不同的化合物对不同肿瘤细胞的抑制作用有所不同,比如:所有受试化合物对Bel-7402,HepG2,U2OS,Bel-7402/5-FU 等细胞株的增殖抑制作用明显,其IC50均为较低的个位微摩尔浓度,抗细胞增殖效果均明显优于阳性对照5-FU,其中,化合物4g 对Bel-7402 细胞的抑制活性最强,其抑制活性是5-FU 的6 倍;对人结肠癌细胞HCT116 和人胃癌细胞HGC27,不同的化合物所呈现的抑制作用差异较大,化合物4f 和4g 活性较弱,其IC50大于12.5 μmol/L,而化合物4a~4e 则表现出与阳性对照药5-FU 相当或更强的抑制活性,说明当连接臂为饱和直链烷烃时,碳链长短对其活性影响较小;当连接臂为不饱和烃基(如化合物4g)或连接臂中含杂原子(如化合物4f)时,抗HCT116和HGC27的活性较差。值得注意的是,所合成的目标化合物对耐药细胞株Bel-7402/5-FU 表现出较强的抑制活性,其抑制活性是5-FU 的37~88 倍,显示较好的抗耐药特性。为了探明目标化合物是否具有选择性抗肿瘤作用,本研究进一步考察了目标化合物对人正常肝细胞LO2的影响。研究发现,1 μmol/L的化合物4a~4g 对LO2 的增殖抑制活性差异较大:化合物4g 和4f 对LO2 几乎没有影响,抑制率分别为3.80%和3.44%;化合物4a 和4c 对LO2 的影响也很小,抑制率小于10%,分别为7.48%和7.12%;而化合物4b,4d 和4e 对LO2 的影响较大,抑制率分别为14.36%,27.61%和22.09%。综上所述,化合物4g 不仅对人肝癌细胞HepG2、Bel-7402,人骨肉瘤细胞U2OS 及耐药细胞Bel-7402/5-FU 表现出强的增殖抑制活性,而且对正常细胞影响很小,表现出较好的选择性,值得进一步研究。
3.3 化合物4g的溶解性及对肿瘤细胞中NO释放的影响
由于大黄酸在水中不溶,在乙醇、丙酮、乙酸乙酯、二氯甲烷等有机溶剂中极难溶解(<0.5 mg/mL),在一定程度上影响其药效的发挥。本研究通过对大黄酸的修饰,以期改善其溶解性能。经测定,体外抗肿瘤活性最好的目标化和物4g 在水中的溶解度与大黄酸相似(<0.5 mg/mL),但在乙醇、丙酮、乙酸乙酯、二氯甲烷等有机溶剂中溶解度较大黄酸有了一定程度的改善:乙醇(>0.1 mg/mL)、丙酮(>1 mg/mL)、乙酸乙酯(>0.33 mg/mL)、二氯甲烷(>2 mg/mL)。
本研究设计的目标化合物是由NO 供体与大黄酸通过连接臂偶联而成,期望得到的化合物能够在体内通过协同效应增强其抗肿瘤活性。为了证实目标化合物抗肿瘤作用是否与NO 释放相关,本研究选取体外抗肿瘤活性最好的化合物4g进行体外NO 释放检测,并用NO 清除剂进行验证。参照文献,采用Griess 法[23]测定了活性目标物4g 对肝癌细胞HepG2 中NO 释放量的影响。经受试药物4g(100 μmol/L)处理后,HepG2 细胞中NO 释放量达20.1 μmol/L。为证明NO 与抗肿瘤活性之间的关系,本研究选用NO 清除剂(血红蛋白)进行验证。实验结果表明,血红蛋白可以显著抑制化合物4g 的细胞毒性(其抑制率由未加清除剂时的80.68%降至44.58%),这一试验结果证明了目标化合物4g的抗肿瘤活性部分来自于NO的释放。
猜你喜欢
中学生数理化·中考版(2021年12期)2021-12-31
中学生数理化·中考版(2021年11期)2021-12-06
载人航天(2021年5期)2021-11-20
食品安全导刊(2021年20期)2021-08-30
昆明医科大学学报(2021年6期)2021-07-31
睿士(2021年5期)2021-05-20
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19
中学生数理化(高中版.高考理化)(2020年2期)2020-11-25
睿士(2020年5期)2020-05-21
睿士(2019年9期)2019-09-10
