
单纯性并/多指(趾)畸形的临床分型及遗传研究进展
2021-05-19 07:20蒲思宇陈静向波江君
妇产与遗传(电子版) 2021年4期
蒲思宇 陈静 向波 江君
并指(趾)畸形(syndactyly)与多指(趾)畸形(polydactyly)是常见的遗传性肢体畸形,发病率高,出生活胎患儿并指发生率为(0.3~1)/1 000,多指为(0.3~3.6)/1 000,男性发病率约为女性两倍[1]。并/多指(趾)畸形表现为手脚指(趾)的畸形,临床外显率差异较大,在家系间和家系内均存在表型差异,上肢较下肢更易受累[2]。其中并指为某些手指和/或脚趾的融合,融合部位可仅为部分指(趾)间皮肤软组织,较重者出现所有指(趾)间的融合、指(趾)骨间骨性融合、甚至掌骨的畸形融合与发育不良。而多指为手和/或脚有其他指(趾),可以是指(趾)远端在纵轴上分叉,也可是骨性/非骨性连接赘生指,重者出现多根指(趾)完全重复伴掌骨、腕骨/跖骨、跗骨的参与[1-3]。并/多指(趾)可以独立出现(单纯型),也可表现在综合征的异常表现中(综合征型)[3]。
本文介绍了单纯性并/多指(趾)的临床表型及当前遗传研究进展,包括并/多指(趾)的分型、遗传模式和已知突变位点,以及引起远端肢体(手脚)畸形的信号通路及相应的调控机制。
一、当前单纯性并/多指(趾)分型及遗传变异
1.并指(趾)
Malik M 等[3]在2012 年对Temtamy-Mckusick 分型扩大延伸,将并指(趾)分为九种,并对各种分型做了详细描述,其内容涵盖了各种表型、基因和分子学的差异。2012年至今,在国内外学者的进一步研究下,将分型进一步完善,见表1。
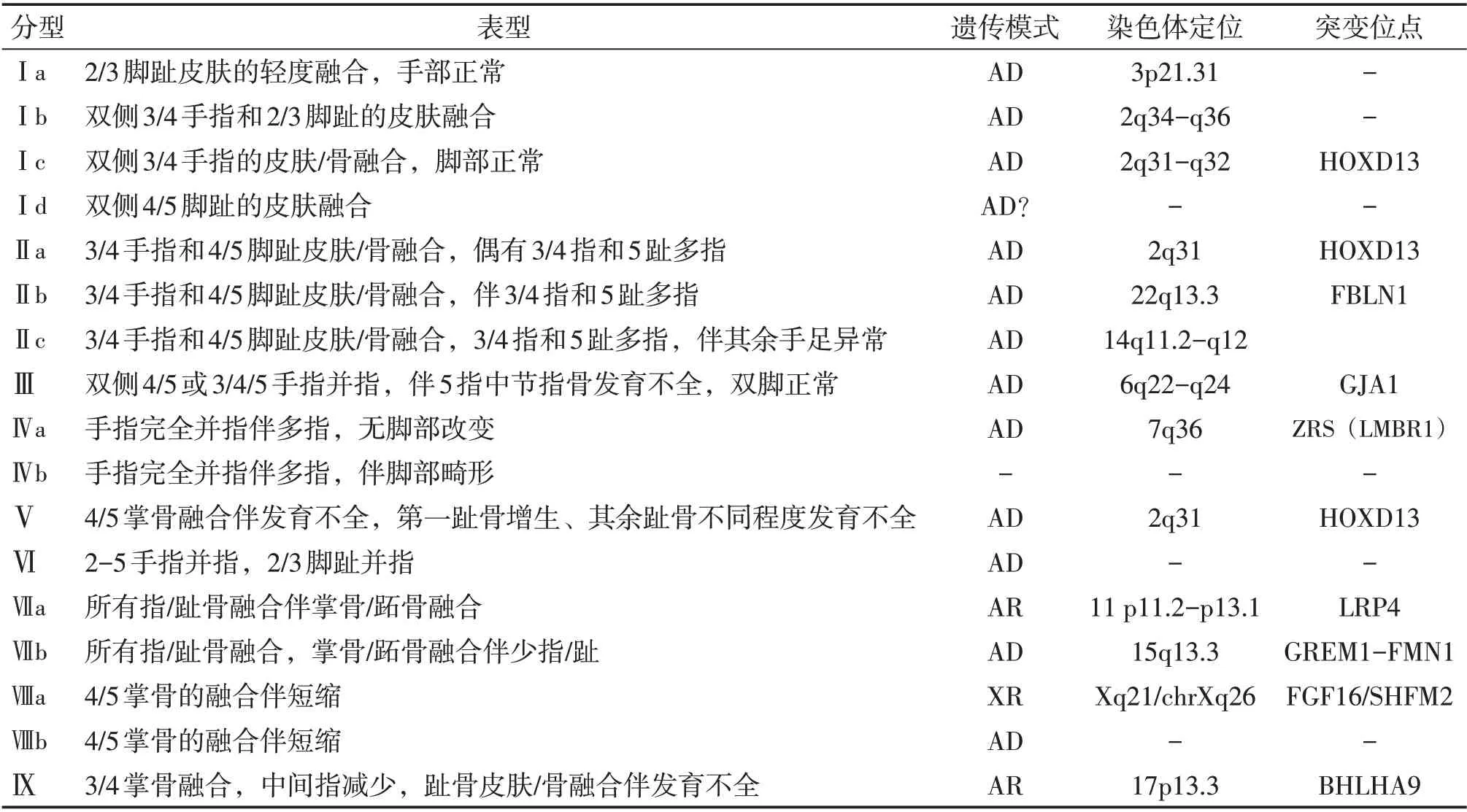
表1 单纯性并指目前分型的对应临床表现与基因突变定位
在各类型并指中,Ⅰ型并指(趾)是最常见的类型,主要表现为中间指(趾)的皮肤融合,即3/4 指和(或)2/3 趾的非骨性并指(趾),涵盖四个亚型;Ⅱ型是唯一合并多指的类型,表现为3/4手指和4/5脚趾皮肤/骨融合,可伴中轴多指或足后轴多趾,根据表型严重程度分为三个亚型;Ⅲ型表现为双侧4/5或3/4/5手指融合伴小指的中节指骨缺失或发育不全,无足部改变;Ⅳ型表现为所有手指的完全融合,伴桡侧或尺侧多指,根据是否合并足部改变分为两个亚型;Ⅴ型的标志是4/5 掌骨融合伴发育不全,可合并短指、并指、屈曲异常、掌纹异常等,足部表现为第一跖骨增生、其余跖骨不同程度发育不全;Ⅵ型表现为对称或不对称的2/3/4/5手指骨与皮肤的完全融合,手外形类似连指手套,合并2/3脚趾的并趾;Ⅶ型类似于Apert综合征,表现为所有指骨难以识别的严重紊乱,骨性融合包括腕骨、掌骨、指骨、甚至桡骨和尺骨,合并缩短和发育不全,常合并下肢同样改变,根据是否少指分为两个亚型;Ⅷ型为4/5 掌骨的融合伴短缩,合并明显的小指尺偏,而无其它异常表现;Ⅸ型表现为3/4 掌骨完全融合后与单根指骨相连,合并中远端指(趾)节发育不良,伴部分非骨性并趾。
此分型除了依据临床表现,还涵盖分子和遗传学差异。在并指(趾)发病因素中,遗传成分起重要作用,大多数的单纯性并指为常染色体显性遗传,但ⅧA型和Ⅸ型为常染色体隐性遗传,而ⅧA为X染色体隐性遗传。基因突变通常以单基因变异为主,目前关于并指(趾)致病基因的研究已经取得了巨大进展,已有多个致病基因得到了确认,包括HOXD13基因、FBLN1基因、GLl3基因、GJA1基因、LRP4基因、GREM1-FMN1基因等[4-6]。
2.多指(趾)
Temtamy-McKusick 根据解剖形态学将多指(趾)分为三个大类,即轴前多指(趾)、轴后多指(趾)和复杂多指,各大类又可细分为数个亚型。2014年Malik S等[1]通过整合每类多指的最新临床和分子进展,对该方案进行了更新,见表2。
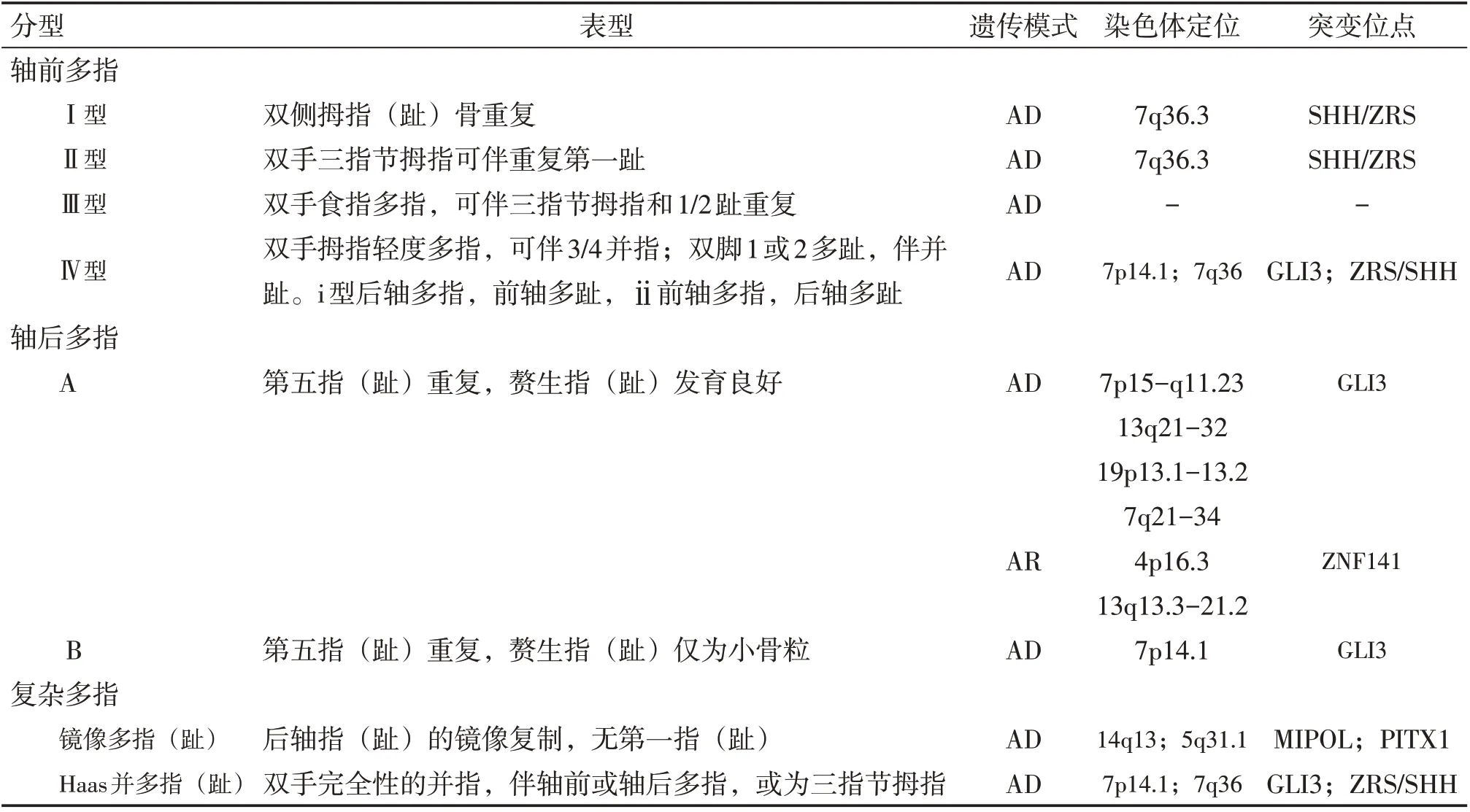
表2 单纯性多指目前分型的对应临床表现与基因突变定位
轴前多指(趾)被分为四个亚型:拇指(趾)多指(Ⅰ型)、三指节拇指(Ⅱ型)、食指多指(Ⅲ型)和手脚差异混合多指(趾)(Ⅳ型),四种亚型可单独存在也可混合出现。目前多项研究表明轴前多指多为常染色体显性遗传,伴有外显率的降低[7]。其中Ⅰ型拇指(趾)多指是最常见的类型,主要表现为拇指(趾)骨重复,轻度仅为末节指(趾)骨变宽(鸭嘴状外观)或末节指(趾)骨重复,重度者复制整个拇指(趾),包括一个掌(跖)骨和两节指(趾)骨;Ⅱ型三指节拇指常双侧对称存在,表现为拇指有一个额外的中节指骨,常有异常长与纤细的第一掌骨,在脚上可伴重复的大脚趾;Ⅲ型食指多指,除食指的重复外合并三指节拇指,有时伴有第一、二脚趾的多指;Ⅳ型手脚差异混合多指(趾)表现为手脚多指的轴向不一致,Ⅰ型轴后多指,轴前多趾,Ⅱ型与之相反。Ⅳ型临床表现手部多指程度轻,表现为末节指骨增宽、分叉和偏斜,偶有不同程度的第三、四指并指,足部第1 或2 趾多指,第一跖骨缩短,胫骨偏斜,可伴有第二、三趾(或所有趾)的并趾。
轴后多指(趾)表现与轴前多指(趾)相反,为尺侧指(趾)的重复表现,根据严重程度分为AB 两组,遗传模式和外显率也有区别。轴后A 型赘生指(趾)发育良好,拥有1 ~ 3 节完整的指(趾)骨,指甲和皮肤皱褶,与第五或额外掌(跖)骨呈关节连接。轴后A 型可根据不同突变位点再分为6个亚型,遗传模式可为常染色体显性或隐性[1,8]。轴后B 型是最常见的类型之一,赘生指(趾)发育不良,仅有小指尺侧/第五趾腓侧软组织突起或小骨粒附着,与正常部分呈非骨性连接,外显率约为43%[9]。虽然目前认为A 型和B 型是遗传异质性的两种类型,但有报道发现在同一家系甚至个体中同时存在A、B 两型,因此这两种是否确为遗传异质性尚未被证实[8]。
复杂多指包含不能归纳于常见轴前或轴后表型的多指(趾),例如镜像多指(趾),Haas型并多指(趾),中央多指(趾)和非轴线多指(趾)等。Haas 型并多指(趾)又被称作Ⅳ型并指(趾),极为罕见,约为1/300 000,只在文献中零星报道[10]。临床表现为双手所有手指完全性的并指,伴有多指表现,多指可能是掌骨加指骨的完全多指,或为三指节拇指。指骨可能融为一个骨团,但是掌骨间不存在骨性联结。指骨融合后向内弯曲,使整个手呈杯状。指甲也常完全融合,或仅有一凹槽作为分界。根据有无脚部的改变将Ⅳ型并指(趾)分为两个亚型,Ⅳa 型(典型Haas 型)患者脚部无异常,而Ⅳb 型(Andersen-Hansen 型)伴有脚趾的异常改变,可为并趾或多趾[3]。
虽然各分型的表型不同,但目前研究发现部分不同表型多指(趾)拥有相似的遗传突变基础,轴前多指、部分轴后多指及Haas型并多指(趾)皆与ZRS/Shh 及Gli 突变有关[7,11]。但大多数多指表型存在其他的遗传变异,特别是复杂多指存在显著遗传异质性,多指(趾)畸形病因中涉及的遗传因素仍有待进一步确定[12]。
二、指(趾)的形成与调控通路
并/多指(趾)的异常表型来源于遗传突变,各突变影响着胚胎早期肢芽的正常发育。胎儿的手脚发育是一种图式发育过程,包含三个轴的发育:远近轴、前后轴、背腹轴。在胚胎早期细胞在三个轴上增殖、分化,第4周末聚集成肢芽,肢芽逐渐增长增粗,并分化出骨、肌肉、皮肤等,形成初步的四肢,至第7~8周,蹼膜消失,形成具有特定外形和数目的手足[13]。其中顶端外胚层嵴(apical ectodermal ridge, AER) 和极性决定区(zone of polarizing activity,ZPA)是肢芽形成的主要形态发生源,两者相互依存[14]。
1.顶端外胚层嵴与信号调节通路
AER由中胚层的肌肉、骨骼前体细胞增殖覆盖外胚层形成,位于肢芽远端边缘背腹交界处,是肢芽上重要的生长信号中心,决定远近轴极性形成,它的缺失会导致远端肢体骨性缺失[15]。AER的形成和发育受维甲酸、成纤维细胞生长因子(fibroblast growth factor,FGF) 和骨形态发生蛋白(bone morphogenetic protein,BMP)调控,其中指(趾)间细胞凋亡是由FGF 和BMP 共同作用于AER 的结果[16]。
AER 三个调控因子中,维甲酸拮抗FGF 信号,是前肢出芽的必需条件[17]。而FGF编码肢芽生长所需要的AER特异性活性因子,家族内有四个成员在AER区域内表达,包括Fgf4、8、9和17,其中Fgf8作用范围大、时间长,贯穿AER形成到凋亡的全过程,信号失活会导致Shh 表达的延迟和肢芽显著减小[18]。BMP作用于肢体发育的各种不同方面,包括手指形态确定、肢体的前后轴和背腹轴的形成以及AER调节。BMP控制指(趾)间组织的凋亡,防止并指(趾)的出现,作用方式包括直接和间接作用[19]。正常的凋亡首先需要BMP介导细胞凋亡信号作用于AER,在肢体发育后期Bmp2、4和7在指间表达上调,提供凋亡信号[20-21]。其次BMP信号传递到AER 区域使区域内Fgf基因表达下调,而Fgf基因表达的下调使指(趾)间组织的细胞存活活性降低,任何一个条件的缺失都会导致并指发生[18]。
2.极性决定区与信号调节通路
(1)ZRS/SHH调节通路
ZPA位于肢芽后部,决定前后轴极性形成,即从第一到第五指(趾)的方向,调控手足的掌骨跖骨及指(趾)的数量与外形形成[21]。在发育过程中,信号分子的不同浓度聚集梯度提供信息、指导发育,这种信号分子被称为形态发生素,而Shh基因的转录产物即为ZPA 的形态发生素[14]。Shh基因定位于人7号染色体长臂远端(7q36),Shh蛋白表达于脊索、背神经管底板、大脑、肠和肢芽的ZPA,参与肢体、神经管及消化道的发育[22]。Shh蛋白经过剪切加工后形成含有C端和N端的多聚体而拥有活性,其C 端有分子内胆固醇转移酶活性,N 端可与胆固醇共价结合形成复合物。Shh的表达具有空间特异性,它在肢芽前部的异位信号是导致多指的主要原因,而Shh的缺失也会导致手的完全缺失和脚仅余一根脚趾[23]。
Shh 信号通路受多种因子调节,其中最重要的是ZPA 调控序列(ZPA regulation sequence,ZRS)。ZRS 也被称为哺乳动物-鱼类-保守序列-1(mammals-fishes-conserved-sequence-1;MFCS-1),长度约800 bp,在物种间高度保守,定位于人染色体7q36.3 上的LMBR1基因的第5 内含子内,位于Shh 基因上游约1 Mb 处,为Shh 基因表达的肢体特异性增强子,它的突变会引起Shh 的异位表达,导致肢体畸形[24-25]。ZRS 序列可以被分成两个区域:位于5’端的活性空间结构域和没有独立活性的3’端结构域,但3’端的信息沿着ZRS传递,影响5’端增强子与Shh 启动子的结合,也通过对整个远程作用的调控来保持长期活性[25]。
(2)ZRS相关综合征
ZRS基因上单个核苷酸突变或者结构改变导致的多种远端肢体畸形被称为“ZRS 相关综合征”,目前已有报道与多种发育畸形联系密切,外在畸形表现均累积手指[10]。根据ZRS突变类型,可划分为点突变型和重复型,虽然突变的位点和序列重复的长度不同,但却有相似的临床表现[7]。目前已报道点突变位点有105C>G,295T>C,305A>T,323T>C,396C>T,446T>A等,表型为三指节拇指多指和II 型轴前多指,而404G>A,404G>C 改变将导致Werner Mesomelic 综合征,除手指表型外还合并足部的多趾和胫腓骨发育不良[26-27]。重复突变的序列长度不定,可为ZRS 序列内的小片段重复或包含ZRS 区域在内的长段重复,临床体征手部均受累,常表现为含三指节拇指的复杂型多指,Ⅳ型并指也被报道与ZRS 序列长段重复有关[10]。Lohan S 等的研究发现ZRS区域内小段的微重复(<80 kb)会导致严重的Laurin-Sandrow 综合征,表现为手足镜面多指畸形、桡骨胫骨缺如、尺骨腓骨重复伴鼻部缺陷。而Andersen-Hansen 型并指虽被报道可能也与ZRS区域的基因重复有关,但目前尚未明确[28]。目前尚无报道人类ZRS序列的缺失,Sagai T等在小鼠实验中发现敲除ZRS 段序列将会导致小鼠Shh 表达完全丧失,近端肢体正常,但肱骨远端和膝关节远端肢体变形缺失[24]。Kvon EZ 等也通过对不同进化阶段的蛇的基因测序,发现残存有骨盆与不成熟后肢的基础阶段蛇与蜥蜴的ZRS 序列有80%的同一性,而完全失去四肢骨骼结构的高级阶段蛇的ZRS序列被大量替代,完全丢失了对ZPA的特异活性[29]。
(3)与ZRS协同调节Shh表达的因子
因为Shh-ZRS在肢芽发育上的重要作用,其详细的调节机制一直是研究热点,目前认为ZRS与多种调节因子协同调控Shh基因转录活性来发挥作用,例如ETS转录因子(E26 transformation-specific)、核不均一核糖核蛋白(heterogeneous nuclear ribonu⁃cleoprotein,HnRNPs)家族等[30]。
ETS 家族蛋白在Shh 的表达空间特异性调控中发挥重要作用,既将Shh 的表达限定在肢芽后缘,同时抑制了其在前缘的异位表达。ZRS 上的ETS 因子结合位点与其他因子结合位点,共同赋予ZRS精确决定Shh时空特异表达的属性[31]。Lettice LA等报道GABPα、ETS1 与ZRS 结合激活Shh基因的转录,而ETV4、ETV5与ZRS结合将会抑制转录,两种效应共同介导Shh 的差异效应,从而确定其空间分布的边界及表达模式[30]。ZRS 的突变导致ETS 因子结合位点数量的改变,将导致Shh 表达异常,引起畸形发生。
HnRNPs 主要与RNA 聚合酶Ⅱ转录复合体结合为hnRNP-RNA 复合物,参与mRNA 的合成、翻译与降解过程[32]。与ZRS特异性结合的蛋白中,有多个蛋白质来自于HnRNPs家族,其中HnRNP K为家族中研究最为广泛的成员,其具有序列特异性结合活性,能与单链或双链DNA 相互作用。Xu C 等的体外实验证实HnRNP K 与ZRS 及SHH 启动子结合,HnRNP K 的缺失不仅抑制了SHH 启动子的活性,而且完全抑制了野生型或突变型ZRS对SHH启动子的增强作用;而ZRS 发生突变,增强其与HnRNP K的结合亲和力,将导致Shh基因的表达上调[33]。
3.其他参与肢体发育的调控信号
AER与ZPA区域细胞的增殖、分化、迁移、凋亡过程除了上述信号通路外,还由多种级联信号之间的协调互作来调节,例如HOX基因、Gli基因、HAND2基因及其相应的信号通路。
HOX基因根据染色体定位不同分为四个基因族群(HOXA、HOXB、HOXC 与HOXD),各族群又可分成13个平行同源组,按照线性排列[34]。处于不同线性位置的基因按照时间和空间发挥不同功能,例如靠近3’端的HOX基因更早表达,并且表达部位靠前,而靠近5’端的基因则相反[35]。HOX基因参与远近轴和前后轴的调控,HOX8-11参与AER的形成和维持,促进肢体细胞的增殖分化,而HOX12、13发挥抑制作用,过量表达可使肢体远端增长提前终止,故在AER 形成早期HOX12 和13 的转录活性低[36]。HOX基因主要通过与Shh基因协同表达参与ZPA 诱导,涉及HOX10-13。早期HOX基因簇按照线性排列顺序依次表达,随后Shh信号将影响HOX基因在发育上的晚期表达,特别是通过将表达域移向后方,调控指(趾)的形态发生[37]。HOX基因表达异常,包括Hoxd 11-Hoxd 13 基因在早期前肢芽中表达会诱导Shh 的镜像表达,导致重复肢体;又例如HoxA 和HoxD 簇的完全缺失会导致shh 表达的缺失,致使远端肢体缺失[38]。
Gli 是Shh信号下游的效应因子,与Shh形成调控胚胎发育的重要调节通路,与指(趾)的发育也密切相关,异常表达常导致多指或并指的发生[39]。1987 年Kinzler 等首次在恶性神经胶质瘤中克隆出GLI1基因,其后GLI家族三位成员依次被发现,三个基因在进化上高度保守。GLI1、GLI2、GLI3均参与远端肢体的图示形成,但GLI3起主要作用。GLI3基因表达产物经过分解后可形成GLI3R亚型,该亚型转运到细胞核负性调节SHH 靶基因的表达。而Shh 信号的激活将抑制GLI3R 亚型的产生,并促进GLI 激活物(GLI2A 和GLI3A)的形成,后者上调Shh靶基因转录水平。因此,在肢芽发育过程中GLl3 和Shh相互制约,以保证正常的远端肢体形成[40]。此外,早期肢芽前后轴极性的形成和肱骨/股骨的特化也是由GLI3 和HAND2 转录因子之间的相互作用介导的,还有研究表明GLI3也与HOXD12基因及SOXB1 蛋白等因子相互作用,参与肢芽的调控[41-42]。
碱性螺旋-环-螺旋转录因子(Hand2)与多种器官发育密切相关,包括心脏及颅面器官,同时也决定早期肢芽的形成[43]。Charité J 等报道Hand2 缺陷的小鼠肢芽间质细胞在胚胎早期大量凋亡,肢芽停止发育,并且无Shh 表达[44]。随后研究发现Hand2 位于Shh 信号上游,含有Hand2 的染色质复合体与ZRS结合,促进ZRS的表达增强活性,激活Shh 表达,并且在肢芽中可通过与Hoxd13 蛋白(Shh抑制因子)结合形成转录复合物而使Shh表达增强。Hand2 在肢芽形成早期完全缺失会破坏Shh在肢芽后部表达域的建立;而不完全或晚期失活,Shh 都将继续表达,但后期Hand2 也仍有助于Shh表达的转录上调[42,45]。
三、目前尚待进一步研究的问题
虽然目前肢体的发育和指(趾)畸形的研究已经越发深入,也解决了诸多疑问,但是仍存在一些重要科学问题尚待解决。首先,大多数分型的并多指(趾)已发现其致病突变,但仍有部分类型没有找到具体的突变位点,如并指(趾)的Ⅰd、Ⅳb、Ⅵ、Ⅷb 等型。其次,指(趾)发育畸形也存在着遗传异质性,例如ZRS/Shh 突变不能解释所有孤立的TPT(--)家族,也不能解释不同类型的TPT(--)表型,应该存在其他机制调控PPD(--)。这都需要进一步的研究来揭示相关调控基因及作用机制。
近日笔者在临床收治了一例Haas 并多指家系,出现表型者为爷爷、父亲和儿子,父亲与儿子皆表现为杯状手伴脚1-3 并趾,爷爷为双手2-4 指并指伴右脚1/2 并趾,其余家庭成员未见异常。对家系三代成员共10人外周血样进行全基因组测序,将患者与正常亲属测序结果进行对比,通过筛查与过滤,在儿子和父亲测序结果中发现在7 号染色体7q36.3区域LMBR1基因内部ZRS序列缺失,且其附近发生染色体内倒位和重复突变。此突变区域在哺乳动物中高度保守,目前未有报道定义该结构变异区域的功能和作用机制,但从患者表型来推测可能参与Shh 基因的表达调控,但具体作用机制是什么?与ZRS增强子序列是否有关仍需进一步的研究。
四、总结与展望
人的上肢功能,特别是手指功能,在全身功能中占重要比重。手的动作精细复杂,且其正常动作很大部分取决于掌弓的完整性和可活动度,任何导致掌弓破坏的疾病都会对手功能产生重大影响。而并/多指,特别是程度严重的类型使掌弓结构变形,腕掌关节、掌指关节、近端指间关节、远端指间关节自由度完全丧失,手指的握、抓、捏等基本功能均无法展现,严重影响患者的日常生活、信息沟通、情感交流和工作能力,给家庭和社会造成沉重的负担。
然而并/多指(趾)畸形患儿在产检时通过常规影像学检查难以发现,而出生后患儿及家庭将遭受来自各方面的压力,严重影响患者生理和心理发育,特别是严重表型者。而在并/多指(趾)发病因素中,遗传成分起重要作用,对其致病突变基因的发现及机制研究,有助于提高孕期筛查检出的可能性,将有效降低严重表型指(趾)畸形胎儿的出生率。并且对并/多指(趾)的基因突变研究可帮助区别单纯并/多指(趾)和各种复杂综合征中的并/多指(趾)表现,对患儿的诊断和后续治疗有重要的意义。
除了对临床的帮助,对并/多指(趾)分子机制的研究同样有助于相关学科发展。哺乳动物肢体的三个轴线都在手上显著体现:由腕到手指对应远近轴,由拇指到小指对应前后轴,手背手掌对应背腹轴。因此对并/多指(趾)的研究将增进对肢体胚胎发育过程的了解,并/多指(趾)基因的确认不仅可以解释指(趾)发育机制和肢芽图示发育模式,还有助于弄清复杂的发育调控模式[3]。并且由于并/多指的遗传异质性和家族间等位基因异质性,故并/多指也是研究发育遗传学的一个非常重要的模型。
总之,本篇综述介绍了目前单纯性并/多指的临床分型及遗传机制,也更深入的总结了远端肢体发育调控机制的研究进展,希望能有助于研究者们更加地关注并/多指及远端肢体发育畸形,继续解决目前仍存在的问题,帮助孕期胎儿筛查及高危家庭患病预防,促进优生优育。
猜你喜欢
中华医学图书情报杂志(2022年1期)2022-11-18
中华骨与关节外科杂志(2022年1期)2022-08-31
中国现代医生(2022年21期)2022-08-22
锦州医科大学报(2022年2期)2022-05-07
医学食疗与健康(2022年2期)2022-04-23
农村科学实验(2022年2期)2022-03-12
昆明医科大学学报(2022年1期)2022-02-28
三农资讯半月报(2020年2期)2020-03-09
学校教育研究(2019年19期)2019-11-23
小天使·二年级语数英综合(2017年10期)2017-10-31
