
三氯化硼离子态光电子谱的理论研究
2021-10-23 06:24宁小寒于同坡陈燕单宝坤周晓国刘世林
量子电子学报 2021年5期
宁小寒,于同坡,陈燕,单宝坤,周晓国*,刘世林
(1 中国科学技术大学,合肥微尺度物质科学国家研究中心,化学物理系,安徽 合肥 230026;2 中国计量科学研究院,北京 100013)
0 引言
三氯化硼(BCl3)作为一种重要的刻蚀气体,主要用作半导体硅的掺杂源,在半导体工业中发挥着重要的作用[1-5]。考虑到等离子体刻蚀或光刻蚀过程中,在大量低能电子轰击或紫外光辐射作用下,中性的BCl3气体分子会发生电离和解离过程,其解离速率和产物性质对于稳定和控制刻蚀过程有着重要的影响。因此,BCl3分子的电离和解离性质的研究,尤其是其离子结构和性质,对于工业应用具有重要的参考价值。
1 理论计算
为了准确描述BCl3电离-解离过程中的结构变化,选取了三种电子密度泛函(DFT)理论结合aug-cc-PVTZ 基组,对BCl3中性分子和基电子态分别进行了结构优化和振动频率分析。此外,针对的电子激发态,采用了同等水平下的含时DFT(TD-DFT)方法完成了结构优化和频率分析(鉴于自然界中B、Cl 同位素分布情况,计算过程使用归一后占比最高即50%的11B、35Cl 构成的11B35Cl3进行后续计算)。此外,考虑到电离过程中分子对称性的剧烈变化,在离子结构优化过程中分析了相应电子态的波函数,以确保轨道对称性的一致性和电子组态的连贯性[20]。
考虑到BCl3的光电离-解离过程中可能涉及到较为复杂的电子相互作用,在实际DFT 计算中选择了有代表性的三种泛函,比较不同泛函中Hartree-Fock(HF)成分差异对分子(离子)结构、频率和能量的影响,分别是拥有较低HF 成分的PBE0[15](25%)、较高HF 成分的M06-2X[16](54%),以及范围分离泛函ωB97XD[17](近程22.2%,远程100%,ω=0.2)。所有的几何优化和振动频率计算均在Gaussian 16[18]程序包中完成。
基于优化的BCl3中性分子、基态态和态结构和振动频率,各离子电子态对应的绝热电离能为
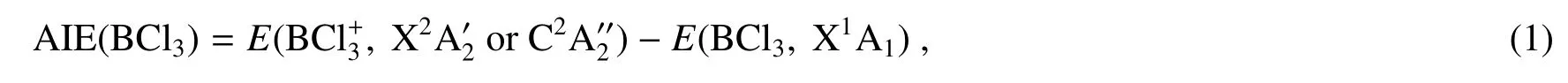
2 结果与分析
2.1 BCl3 中性分子和和态的优化构型
图1 显示了BCl3中性分子主要前线分子轨道。显然,BCl3中性分子的HOMO、HOMO-1、HOMO-2、HOMO-3 和HOMO-4 电子主要为Cl 原子的孤对电子形成的非键轨道,而HOMO-5 则为B-Cl 大π 键的贡献,LUMO 对应B-Cl 反π 键,这与以往的计算结果[21]完全一致。考虑到能量的近简并关系,、和B2E′态的生成分别来源于HOMO、HOMO-1/HOMO-2、HOMO-3/HOMO-4 孤对电子的剥离,这很好地解释了这些低离子态的轨道成分[10,11,13],同时,不同Cl 原子的p轨道孤对电子的失去也引起了分子对称性的坍塌。而相对更高的离子态的生成则来源于HOMO-5 电子的剥离。显然,对态而言,大π 键电子的减少显著削弱了B-Cl 键,从而导致其三个B-Cl 键长同时拉长至0.1795 nm,同时保持键角皆为120°的平面构型。

图1 BCl3 中性分子前线分子轨道示意图Fig.1 Schematic diagram of the frontier molecular orbitals of neutral BCl3 molecule
表1 不同计算水平下得到的BCl3 分子、 和态结构参数aTable 1 Geometry parameters of the BCl3 neutral and the in and state optimized at different levels of theory

表1 不同计算水平下得到的BCl3 分子、 和态结构参数aTable 1 Geometry parameters of the BCl3 neutral and the in and state optimized at different levels of theory
a:Using the aug-cc-PVTZ basis set,with the units of bond length(nm)and bond angle(degree);b:Ref.14.
相对BCl3中性分子而言,基态表现出明显的1S2L 特征,B-Cl(1)键长由0.1740 nm 缩短至0.1665 nm,其它两个B-Cl 键则略微加长,同时,平面内键角也发生较大变化。考虑到如此明显的分子结构变化,BCl3分子电离时必然伴随着强烈的B-Cl 非全对称伸缩振动和弯曲振动激发,并且电子能谱中基态谱带中最强振动跃迁应偏离0-0 振动。这在以往的实验光谱[14]中已经得到了证实。
采用PBE0/aug-cc-PVTZ 水平下优化的离子构型,在相同理论水平下分别计算得到了和态的振动频率(见表2)。其中,态6 个振动模式分别对应2 个面内弯曲振动模式1 个面外弯曲振动模式、1 个全对称伸缩模式以及2 个非全对称伸缩模式其振动频率分别是191、225、383、482、699、1114 cm-1,这些结果与Yang 等[14]的CCSD(T)/aug-cc-pVTZ 计算结果和PFI-PES 实验频率基本吻合。相对而言离子态与中性分子基态对称性相同(D3h点群),2 个弯曲振动(或)为二重简并模式,振动频率为223 cm-1,e′对称性;面外弯曲振动振动频率为428 cm-1,对称性;B-Cl 键的对称伸缩振动振动频率为442 cm-1,对称性;2个非全对称伸缩振动(或)为二重简并模式,振动频率937 cm-1,e′对称性,各振动矢量如图2 所示。

图2 BCl3 分子和离子3 个B-Cl 伸缩振动相关的振动模式,其中黄色箭头表示振动矢量方向Fig.2 Three B-Cl stretching vibrational modes of the BCl3 neutral and cation,where the yellow arrows represent the vibrational displacement vectors of atoms
在表2 的振动频率基础上,结合离子几何结构以及各振动模式矢量,计算了300 K 温度下BCl3跃迁对应的Franck-Condon 因子。表3 列举了计算得到的主要振动谱峰位置、Franck-Condon 因子和振动标识。显然,对的光电子能谱(PES 或TPES)而言,0-0 振动跃迁不具有最大的跃迁强度,主要的振动跃迁对应B-Cl 全对称伸缩振动序列,且低振动量子数的振动跃迁强度明显处于主导地位。这些计算结果清楚地表明了BCl3电离生成离子时将主要伴随B-Cl 全对称伸缩振动激发,与其分子结构(主要是键长)的变化趋势完全一致。
表2 和态振动模式和计算频率Table 2 The vibrational model and frequencies of the ground state of and the electronic excited state of

表2 和态振动模式和计算频率Table 2 The vibrational model and frequencies of the ground state of and the electronic excited state of
a:At the PBE0/aug-cc-PVTZ level,with unscaled frequencies;b:From Ref.14.
表3 300 K 时,BCl3(X1A1)→跃迁谱带中主要的振动跃迁位置、Franck-Condon 因子,以及振动标识Table 3 The main vibrational transition positions,Franck-Condon factors and the vibrational assignments of the BCl3(X1A1)→transition at 300 K

表3 300 K 时,BCl3(X1A1)→跃迁谱带中主要的振动跃迁位置、Franck-Condon 因子,以及振动标识Table 3 The main vibrational transition positions,Franck-Condon factors and the vibrational assignments of the BCl3(X1A1)→transition at 300 K
a:Experimental data from Ref.11;b:Calculated at the PBE0/aug-cc-PVTZ level without anharmonic corrections in the left column,while the right lists the results after a blue shift by 0.583 eV;c:Deviations of the calculated(after the blue shift)and experimental positions,due to anharmonic effect;d:FC factor is defined as the overlapping integral of the vibrational wave functions between initial state and target state;e:Calculated as the product of FC factors and thermal population.

图3 300 K 时BCl3(X1A1)→跃迁的实验和计算光电子能谱Fig.3 The experimental and Franck-Condon calculated photoelectron spectra of the BCl3(X1A1)→transition at 300 K
由于计算精度的限制,通常理论计算得到的电离能较实验值有较大的偏差,为此,在做光谱拟合时,常需要将计算的谱带适当能量平移。如表4 所示,采用三种密度泛函计算的各电子态对应的电离能与已有的CCSD(T)计算结果[14]和实验值[10,11,13]基本一致,但的确存在一定的偏差。以基态态为例,在不同水平下得到的基态态绝热电离能AIE 的计算结果,相对公认为黄金标准的CCSD(T)结果[14]和实验值[14]偏低0.12~0.38 eV。鉴于这些密度泛函方法和CCSD(T)在计算分子结构(包括中性分子和离子和态)方面结果基本一致(见表1),同时密度泛函计算较CCSD(T)具有明显的经济优越性,这样的能量计算误差尚可接受。因此,此处依然选择PBE0/aug-cc-PVTZ 水平下得到的态计算结果做光谱模拟。
表4 BCl3 电离得到 和态离子态的绝热电离能(AIE)和垂直电离能(VIE)Table 4 The AIE and VIE values of the transition BCl3 (X1A1)→( or )

表4 BCl3 电离得到 和态离子态的绝热电离能(AIE)和垂直电离能(VIE)Table 4 The AIE and VIE values of the transition BCl3 (X1A1)→( or )
a:From Ref.10;b:From Ref.11;c:From Ref.13;d:The present results based on Franck-Condon simulation and calculated at the PBE0/aug-cc-PVTZ level considering ZPE corrections;e:From Ref.14;f:Present calculation results with ZPE corrections.
实际模拟中,不仅将计算的谱带适当蓝移(0.583 eV),而且考虑到仪器分辨和谱线加宽的影响,对每个振动激发(图3 中的棍状图)采用了28 meV FWHM 的高斯线型处理,最终叠加获得的态Franck-Condon 跃迁的光电子能谱,如图3 蓝色曲线所示。显然,Franck-Condon 拟合的光谱与实验谱图[11]吻合得很好。值得注意的是,实验观测的第一个明显的振动谱峰位于14.240 eV 附近(即谱峰阈值,图3 中星号标记),并不对应0-0 跃迁,而是振动热带的贡献,即BCl3中性分子基态X1A1的B-Cl 全对称伸缩振动(ν4)频率为471 cm-1(58 meV),这与以往的实验值471 cm-1一致[23]。相应地,真正的0-0 跃迁位于第二个振动谱峰(见表3),即14.298 eV 处(图3 中以红色箭头表示)。这样,态的绝热电离能最终确定为14.298 eV。同时,依赖表3 中各振动谱峰的标识和实验能量间隔,可以通过

3 结论
猜你喜欢
无线电工程(2022年10期)2022-10-24
初中生学习指导·中考版(2022年4期)2022-05-12
昆明医科大学学报(2021年8期)2021-08-13
延边大学学报(自然科学版)(2021年1期)2021-04-27
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
中学教学参考·理科版(2016年3期)2017-05-19
中学生数理化·八年级数学人教版(2016年6期)2016-08-22
读写算·小学中年级版(2016年5期)2016-05-14
中学化学(2015年10期)2015-12-14
读写算·教研版(2014年12期)2014-09-01
