
荧光粉中激活剂离子掺杂格位分析
2022-01-23 13:57姬海鹏
发光学报 2022年1期
姬海鹏
(郑州大学 材料科学与工程学院, 河南 郑州 450001)
1 引 言
当前主流白光半导体发光二极管(LED)器件采用蓝光LED芯片复合多色荧光粉方案。大多数荧光粉是某种离子取代型固溶体,固溶体的母体被称为基质,进行掺杂取代的离子常被称作激活剂离子。某一荧光粉是否可用于白光LED,以及所封装光谱转换型白光LED器件的流明效率、相关色温、显色指数等,很大程度上取决于荧光粉的发光特性,因此荧光粉的发光特性研究至关重要。
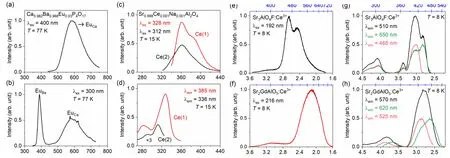
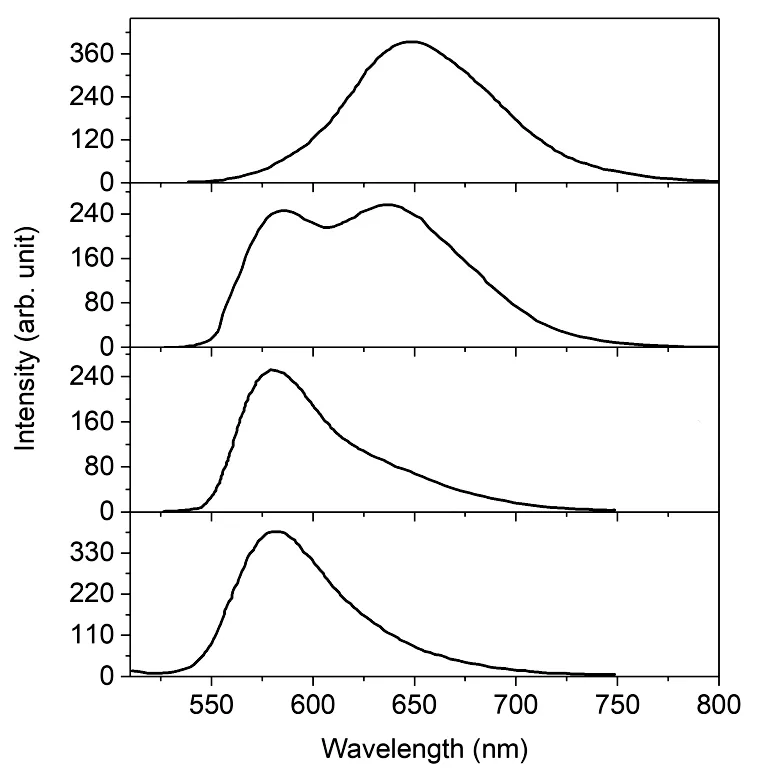
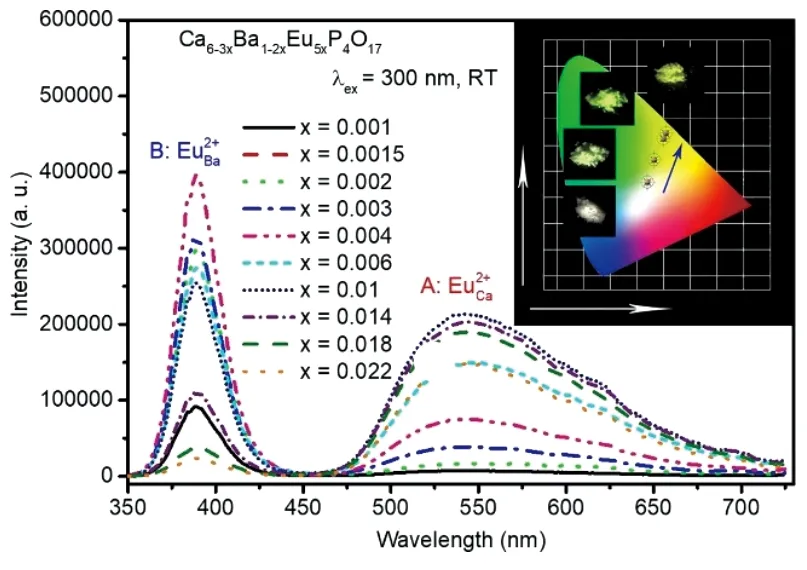
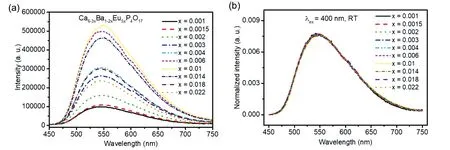
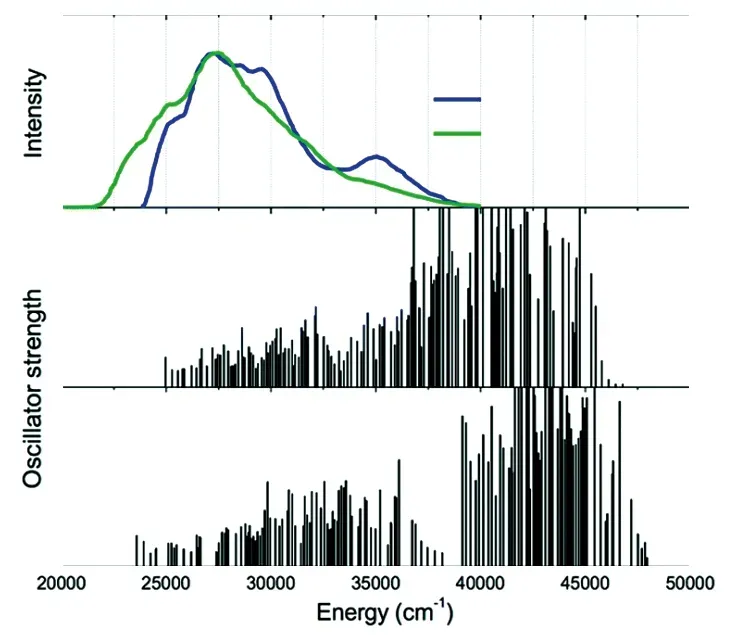
商业化的白光LED荧光粉中所用激活剂离子主要为稀土离子Ce3+、Eu2+与过渡金属离子Mn4+。其中,稀土离子Ce3+和Eu2+的荧光来自于宇称允许的d→f跃迁。该电子构型下,最外层为裸露的5d轨道电子,易受晶体场效应和电子云膨胀效应影响。研究表明,d→f跃迁所受晶体场强度(以εcfs表示)取决于稀土离子在基质中形成的最近邻配位多面体的配位数、平均键长、点群对称性和畸变程度,而其所受电子云膨胀效应的强弱(以εc表示)取决于成键的离子键/共价键性和成键阴离子的极化率[1-2](有兴趣的读者可参考代尔夫特理工大学Dorenbos教授对εcfs和εc的定量计算[3]);因此,Eu2+/Ce3+在不同基质中掺杂时可表现出在发光波长、发光光谱半高宽、量子效率、热猝灭稳定性等方面迥异的荧光性质。比如,在Eu2+/Ce3+掺杂的硫化物或氮化物类基质中更容易得到长波长发光荧光粉,因为此时Eu/Ce原子(电负性分别为1.01/1.06)与S原子或N原子(电负性S 过渡金属离子Mn4+具有3d3电子构型,其荧光来自于宇称禁戒的d→d跃迁。Mn4+离子d轨道在八面体配位晶体场中发生能级劈裂,形成eg轨道和t2g轨道,后者可供Mn4+离子的3个d轨道电子占据。由于正电荷数多,Mn4+常受强晶体场作用,最低激发态为2Eg(2G)能级,其在Tanabe-Sugano图中几乎是一条直线,因此2Eg→4A2g跃迁受晶体场的影响较小。但Mn4+离子的发射跃迁(2Eg→4A2g跃迁)受电子云膨胀效应影响很大,发光能量与Mn4+和配体离子(如F-或O2-)所成键的离子键/共价键性密切相关[11]。2Eg→4A2g跃迁为自旋禁戒跃迁,当Mn4+所处八面体格位存在畸变(如MnOxF6-x构型)而导致点群对称性低于Oh时,电偶极跃迁选律可得到放宽而使Mn4+离子的荧光寿命缩短[11]及得到强零声子线发光和发光峰的劈裂[12]。 综上,Ce3+/Eu2+/Mn4+离子的荧光性质很大程度上取决于其在基质晶格中所形成最近邻配位多面体的特征。了解其所占据格位是了解其所形成最近邻配位多面体特征的第一步,对于理解其构效关系、开发新型荧光粉具有重要意义。目前文献报道采用不同方法来研究激活剂离子的格位占据情况,本文总结了这些方法,将其归为三大类(即光谱学方法、结构分析法和计算光谱学方法),并通过相关研究实例进行对比分析。 2.1.1 激发波长依赖的发光光谱和监测波长依赖的激发光谱 激活剂离子的荧光特性受配位环境影响,因此其在基质中占据具有不同晶体学特征的格位时将分别表现出相应的荧光特性;因此,对该荧光粉而言,改变激发波长将得到激发波长依赖的发射光谱,而改变监测波长将得到监测波长依赖的激发光谱。 梁宏斌等[13]研究了Ca5.982Ba0.988Eu0.03P4O17荧光粉激发波长依赖的发射光谱。如图1(a)所示,当用400 nm激发时,发射光谱中只含有一个半高宽较宽、非对称、主峰位于588 nm的发射带;而改用300 nm激发时,发射光谱中除上述宽发射带外,新出现了一个半高宽较窄、主峰位于389 nm的发射带(图1(b))。在晶体场理论指导下,作者将这两个发射带分别归属于Eu2+占据基质中Ca2+和Ba2+格位时的发光。Shi等[14]通过激发波长依赖的发射光谱和监测波长依赖的激发光谱方法证明Ce3+在SrAl2O4基质中占据两个晶体学格位。如图1(c)所示,改变激发波长后得到了不同的发射光谱:在328 nm激发下,发射光谱中包含两个峰值波长分别为361 nm和384 nm的发光带,来自于Ce3+最低5d1能级到4f能级(2F5/2和2F7/2光谱项)的跃迁(标记为Ce1);而在312 nm激发下,发射光谱中包含一个涵盖320~440 nm、峰值波长分别为336~361 nm发光带,这与Ce1的发光带明显不同,因此推断其来自于占据另一Sr2+格位的Ce3+(标记为Ce2)。如图1(d)所示,当监测不同波长时得到了不同的激发光谱,进一步证明Ce3+同时占据SrAl2O4基质中两个Sr2+格位,其所受晶体场效应明显不同且主要占据Ce1格位(因其发光强度更高)。 图1 (a)~(b)Ca5.982Ba0.988Eu0.03P4O17荧光粉在液氮温度(77 K)及不同激发波长下的发射光谱[13];Sr0.998Ce0.001Na0.001-Al2O4荧光粉在液氦温度(15 K)及不同激发波长下的发射光谱(c)和紫外-可见光区不同监测波长下的激发光谱(d)[14]; 液氦温度(8 K)下Sr2.98Ce0.02AlO4F荧光粉的发射光谱(e)和多种监测波长下的激发光谱(g)以及Sr1.98-Ce0.02GdAlO5的发射光谱(f)和多种监测波长下的激发光谱(h)(为方便对比,将监测其他发光波长处的激发光谱强度设置为监测最强发光峰所得激发光谱强度的一半)[15]。 当基质晶格中含有多种晶体学格位时,激活剂离子是否都占据这些晶体学格位,也可通过激发波长依赖的发光光谱和监测波长依赖的激发光谱方法进行研究。Ba2SiO4中含有两个不同的Ba2+格位,分别与10个或9个O2-配位。为研究Ce3+在Ba2SiO4中的格位占据情况,Lin等[17]测试了Ba1.999Ce0.0005Na0.0005SiO4在不同激发波长下的发光光谱,监测不同发光的激发光谱,示于图2。当掺杂Ce3+后,荧光粉在不同波长紫外光激发下都发出两个峰值分别为375 nm和405 nm的宽带光谱且两个峰值之间的能量差为1 940 cm-1,这与Ce3+离子5d1能级到4f(2F5/2和2F7/2光谱项)跃迁发光特征一致,故推断Ce3+在Ba2SiO4中只占据其中一个Ba2+格位;监测发射光谱中360,376,405,430 nm发光所得系列真空紫外激发光谱可见,所得激发光谱的相对强度与所监测发光波长的强度具有一致性,且各个峰值波长位置一致,佐证了Ce3+离子在该基质中只占据了一种晶体学格位。 图2 Ba1.999Ce0.0005Na0.0005SiO4荧光粉的发射光谱(EM,T=4 K)与真空紫外激发光谱(EX,T=26.5 K)[17] 此外,值得一提的是,还有一些激活剂离子,随基质格位点群对称性或晶体场强度效应不同而表现出相应具有独特特征的荧光光谱,因此根据该荧光光谱特征可推断其所占据格位的点群对称性(包括是否存在反演对称性)或配位体形状。如Eu3+离子的发射光谱来自于5D0能级到7FJ(J=0,1,2,3,4,5,6)能级的f→f跃迁,在其发射光谱中强度最高的谱线分别来自于5D0→7F2电偶极跃迁(613 nm处)和5D0→7F1磁偶极跃迁(596 nm处),两者的相对强度与Eu3+离子所处格位是否具有反演对称性密切相关[18];此外,5D0→7F0跃迁发射峰的数量与Eu3+离子在晶体中所占据的格位数一致,且5D0→7F0发射峰强度较高时说明Eu3+占据的格位非中心对称,而5D0→7F0发射峰观测不到强度时说明Eu3+占据中心对称的晶体学格位。还有,Mn2+离子的发射光谱来自于从4T1到6A1能级的d→d跃迁,该跃迁受制于基质格位的晶体场强度[19]:在四面体配位(弱晶体场)时,Mn2+通常为绿光发光;而在八面体配位(强晶体场)时,Mn2+通常为橙光与红光之间发光。因此,根据Mn2+离子的发光颜色可推断其占据四面体还是八面体格位。 综上所述,通过激发波长依赖的发光光谱和监测波长依赖的激发光谱方法,可以方便地研究激活剂离子在基质中形成的发光中心的种类;此外,对于Eu3+和Mn2+等,还可判定其所占据格位的点群对称性或配位体形状。但当基质中存在多个晶体学格位时,不能明确确定具体占据哪些格位;且在一些情况下,激活剂离子虽只占据一种晶体学格位,但因多种电荷平衡机制而形成多种发光中心。 2.1.2 波长依赖的荧光衰减曲线 激活剂离子受激后从基态跃迁到激发态,停止激发后又从激发态跃迁回基态;荧光粉表现出的荧光强度(统计信息)达到激发时最大强度的1/e时所需时间被称为荧光寿命,而荧光寿命由自发辐射跃迁几率和无辐射跃迁几率共同决定。占据不同格位时激活剂离子荧光特性不同,不仅体现在荧光光谱上,也表现在其荧光衰减曲线和荧光寿命上。因此,可通过测试波长依赖的荧光衰减曲线来分析激活剂离子的格位占据。 如图3所示,笔者曾报道Sr2.98Ce0.02AlO4F和Sr1.98Ce0.02GdAlO5两种同构荧光粉监测不同发光波长的荧光衰减曲线[15]。首先,这些荧光衰减曲线表现出依赖于监测波长的特点;其次,各荧光衰减曲线无法用单指数函数拟合而需要用双指数拟合。如图3所示,采用公式(1)所示的双指数函数进行拟合: 图3 Sr2.98Ce0.02AlO4F(a)和Sr1.98Ce0.02GdAlO5(b)荧光粉在λex=405 nm激发下监测不同发光波长所得室温荧光衰减曲线[15] I(t)=A1exp(-t/τ1)+A2exp(-t/τ2), (1) 其中,I(t)是时间t时的荧光强度,A1和A2是常数,τ1和τ2是荧光衰减时间。 各曲线可用双指数函数很好地拟合,拟合所得τ1和τ2差别较大,分别对应于Ce3+在Sr3AlO4F和Sr2GdAlO5基质中占据8h格位时由于电荷平衡机制不同而形成的两种微观配位多面体。再通过公式(2)计算监测各发光波长时的平均荧光寿命τave: (2) 得到Sr2.98Ce0.02AlO4F荧光粉发射光谱中505,460,550 nm发光的τave分别为38.1,32.5,47.0 ns,而Sr1.98Ce0.02GdAlO5荧光粉发光光谱中570,525,640 nm发光的τave分别为36.1,26.8,42.3 ns。可见,发射较高能量的Ce3+离子微观配位多面体的荧光寿命明显短于发射较低能量的Ce3+离子微观配位多面体的荧光寿命。可能原因是两者之间存在能量转移,或者发射高能量的微观配位多面体的室温热猝灭效应更显著。 2.1.3 时间分辨荧光光谱 荧光粉的荧光强度与其中处于激发态的激活剂离子的数目成正比,荧光强度随时间的衰减速率可反映相应离子在激发态的停留时间。当处于激发态的离子发生无辐射跃迁回基态或发生离子间能量转移时,所测荧光强度的衰减速率将变化,这为研究荧光粉中激活剂离子的格位占据提供了一种手段。 梁宏斌等[13]报道采用时间分辨荧光光谱手段研究Ca6BaP4O17∶Eu2+荧光粉中Eu2+离子所占据格位。如图4(a)所示,406 nm激发下,在延迟时间td=100 ns时,荧光发射光谱呈不对称特征且半高宽相对窄。随着延迟时间td增加为500,1 500,4 020 ns,荧光光谱逐渐宽化(在长波长一侧更加显著)。时间分辨发射光谱的变化说明该发射光谱来自于占据不同阳离子格位而具有不同荧光寿命的Eu2+离子的荧光,即Eu2+同时占据Ca6BaP4O17结构中的两种Ca2+离子格位。在其晶体结构中,Ca(2)—O的平均键长(0.242 0 nm)略短于Ca(1)—O的平均键长(0.251 0 nm),且[Ca(2)O7〗多面体的点群对称性(C1)低于[Ca(1)O8]多面体的点群对称性(Cs),因此将荧光光谱中高能侧发光归因于占据Ca(1)格位的Eu2+,而低能侧发光归因于占据Ca(2)格位的Eu2+。 为进一步明确Eu2+占据Ca(1)和Ca(2)格位的发射峰位置(发射能量),选取td=100 ns和td=4 020 ns两个时间分辨发射光谱进行高斯拟合分峰。如图4(b)所示,两个光谱可分别经高斯拟合分为两个发光峰,峰值分别在2.37 eV和2.18 eV,彼此一致,分别归属于EuCa(1)和EuCa(2)。对于给定的跃迁,荧光衰减时间可认为与其发光波长的三次方成正比[20],因此随着延迟时间td从100 ns增加到4 020 ns,EuCa(1)发光的衰减更快而对整个发光光谱的贡献逐渐减小,解释了图4(a)所示发光光谱逐渐向长波长低能侧宽化的现象。 图4 (a)77 K下Ca5.994Ba0.996Eu0.01P4O17荧光粉在406 nm激发下的归一化时间分辨发射光谱;所选定延迟时间下发射光谱的高斯拟合:(b)td=100 ns,(c)td=4 020 ns[13]。 时间分辨荧光光谱也被用于证明多格位占据的激活剂离子间存在能量传递。Sohn等[21]报道了Sr2Si5N8∶0.0005Eu2+和Sr2Si5N8∶0.02Eu2+荧光粉的时间分辨荧光光谱及其高斯分峰结果(原文在波长坐标下分峰,笔者建议在波数坐标下进行),如图5所示。当掺杂浓度为0.05%时,两个高斯拟合所得发射峰之间的强度比不随延迟时间的变化而变化;而当掺杂浓度增大到2%时,这两个高斯拟合所得发射峰之间的强度比随延迟时间变化而显著不同,证明Eu2+在Sr2Si5N8中占据两个Sr2+离子格位;此外,短波长发射峰的荧光衰减更快而长波长发射峰衰减较慢,可能原因是在高Eu2+掺杂浓度时,占据两个Sr2+格位的Eu2+离子间存在明显的能量传递。 图5 Sr2Si5N8∶0.0005Eu2+(a)和Sr2Si5N8∶0.02Eu2+(b)荧光粉的时间分辨荧光光谱及其高斯分峰结果[21] 2.1.4 变温发射光谱与变温荧光衰减曲线 当激活剂离子占据不同格位且在不同格位表现出不同的热猝灭特征时,根据其荧光随温度升高发生热猝灭行为的不同,可判断激活剂离子所占据格位数。荧光热猝灭本质是无辐射跃迁几率的提高,可表现为荧光强度的减弱或荧光寿命的缩短,因此可分别测试变温发射光谱或变温荧光衰减曲线,以期表征激活剂离子在不同格位的热猝灭行为。 图6(a)所示为Ca5.982Ba0.988Eu0.03P4O17荧光粉归一化的变温发射光谱。在77~500 K温度范围内,随温度升高,长波长发光强度较快发生猝灭而短波长发光强度较慢发生猝灭,使得归一化发光光谱表现出主峰蓝移53 nm的特征。这是因为Eu2+离子占据其晶体结构中的Ca(1)和Ca(2)格位且表现出不同的热猝灭行为。进一步采用变温荧光衰减光谱表征Eu2+在上述两格位的不同热猝灭行为。图6(b)、(c)为在406 nm激发下分别检测500 nm(对应于EuCa(1))和640 nm(对应于EuCa(2))发光在50~430 K温度范围内的变温荧光衰减曲线。随温度升高,热猝灭效应产生,荧光衰减曲线逐渐偏离指数衰减特征[22]。将不同温度下的荧光寿命按照Mott公式进行拟合(分别示于图6(b)、(c)插图中): (3) 其中,τ0为Eu2+在50 K时的荧光寿命,A为常数,Ea为热活化能,k为玻尔兹曼常数(8.6172×10-5eV·K-1),T为温度。可得相应热活化能分别为0.16 eV和0.11 eV,即Eu2+在两个Ca格位中表现出不同的热猝灭行为。因此,根据图6所示变温发光光谱和变温荧光衰减曲线可断定Eu2+占据两个具有不同热猝灭特性的格位中。 图6 Ca5.982Ba0.988Eu0.03P4O17在77~500 K温度范围内归一化的发射光谱(a)、该样品中EuCa(1)(b)和EuCa(2)(c)在50~430 K温度范围内的荧光衰减曲线(插图所示为EuCa(1)和EuCa(2)在不同温度下的荧光寿命及其拟合结果)[13]。 2.1.5 掺杂浓度依赖的发射光谱 当激活剂离子在基质中占据多个格位且其在多格位中的占据倾向性不同或因在某一格位的发射光谱与其在另一格位的激发光谱重叠而可发生能量转移时,随掺杂浓度的逐渐提高,将观察到其在不同格位因浓度猝灭行为不同而表现出的光谱演变;此外,根据低掺杂浓度和高掺杂浓度时荧光光谱的不同,可确定激活剂离子的优先占据格位。 Piao等[23]报道了变Eu2+浓度系列Ba2-xEux-Si5N8氮化物荧光粉的荧光光谱。如图7所示,当x=0.04时,为发射峰值在580 nm的橙光宽带光谱;继续增加Eu2+掺杂浓度,在640 nm处新出现了一个红光发射峰;当x=0.15时,该红光发射峰强度与橙光发光峰强度接近;而当x=0.20时,荧光光谱中只可观察到该红光发射峰。这两个发光峰分别来自于占据Ba2Si5N8结构中两个Ba2+格位的Eu2+离子,且Eu2+优先占据产生较小晶体场劈裂效应的、具有更长成键键长的Ba2+格位。随着掺杂浓度的增加,该Ba2+格位上的Eu2+离子首先发生浓度猝灭,同时把能量传递给产生较大晶体场劈裂效应而发射红光的Ba2+格位上的Eu2+离子。因此,通过该变掺杂浓度发射光谱可知,Eu2+占据基质晶格中两个不同的Ba2+离子格位,且表现出明显的占据倾向性,即优先占据形成较大体积最近邻多面体的Ba2+离子格位。 图7 不同Eu2+浓度掺杂Ba2-xEuxSi5N8的发射光谱[23] 在Ca6BaP4O17∶Eu2+荧光粉中也报道了随Eu2+掺杂浓度增加而变化的荧光光谱。在该基质中Eu2+离子可同时占据Ba2+、Ca(1)2+和Ca(2)2+格位[13],且在这三种格位中占据的倾向性不同,经晶体结构精修,确定其不同Eu掺杂浓度样品的化学组分可写作Ca6-3xBa1-2xEu5xP4O17。 在x= 0.001~0.022范围内制备一系列不同Eu掺杂浓度样品,其荧光光谱示于图8。可以看出,该系列荧光粉的荧光光谱中包含一个位于短波长区的较窄的发射带和一个位于长波长区的较宽的发射带;随着Eu2+掺杂浓度的增加,两个发射带的演变行为有显著差异。在x=0.001时,高能发射带强度很高而低能发射带强度很低;随着Eu2+掺杂浓度逐渐增加,两个发射带强度都有所提高;之后,高能发射带强度降低,低能发射带强度增加并超越高能发射带强度(得益于占据低能发射带晶体学格位的Eu2+数量的增加和来自于高能发射带的能量转移);最后,由于浓度猝灭效应,继续提高Eu2+浓度使得两个发射带的强度都有所降低。因此,当激活剂离子在不同掺杂格位表现不同的掺杂倾向性时,可从其掺杂浓度依赖的发射光谱中观察到显著不同的浓度猝灭现象。 但当激活剂离子在多个格位中的掺杂倾向性一致或非常接近时,其在多个格位中的浓度猝灭现象将几乎一样而无法分辨。实际上,图8所示的低能侧较宽的发射带来自于同时占据Ca(1)和Ca(2)两个格位的Eu2+离子[13]。晶体结构精修结果表明,不同浓度掺杂时Eu2+离子在这两个Ca2+格位的占位率一致。因此,如图9所示,该低能发射带随着Eu2+掺杂浓度的增加虽然表现出有规律的浓度猝灭效应,但从其归一化光谱可见,半高宽和峰值波长都没有发生变化,表现出与Eu2+占据单一格位时相同的演变行为。因此,当激活剂离子占据多个格位但其占位倾向性一致时,难以通过浓度猝灭效应来判断其是单一格位占据还是多格位占据。 图8 Ca6-3xBa1-2xEu5xP4O17的室温发射光谱(插图所示为其中4个掺杂浓度样品在300 nm激发下的实物照片和相应的CIE坐标图)[13] 图9 Ca6-3xBa1-2xEu5xP4O17的室温发射光谱(a)和归一化发射光谱(b)[13] 2.2.1 晶体结构精修 很多荧光粉都是一种离子掺杂型固溶体;激活剂离子掺杂入基质晶格后,由于其具有与被取代离子不同的离子半径和相对原子质量,且对入射X射线的衍射能力也不同,因此将造成掺杂后荧光粉的X射线衍射谱(XRD)与未掺杂基质物相的XRD有所不同。多数情况下,实验所合成荧光粉为多晶粉体,利用粉晶结构精修方法如Rietveld方法,可将激活剂离子分别放入不同的可能晶体学掺杂格位,通过比较其理论所得XRD与实测XRD之间的异同而确定激活剂离子在不同格位的掺杂占据情况。 相对于绝大多数基质中所含离子而言,稀土激活剂离子Ce3+和Eu2+具有更大的相对原子质量,为通过Rietveld等晶体结构精修过程分析Ce3+/Eu2+在荧光粉中的格位占据及占位率提供了有利条件;但受浓度猝灭效应制约,荧光粉中激活剂离子的掺杂浓度一般不高,使得通过晶体结构精修方法精确分析激活剂离子的格位占据和占位率具有一定困难。因此,在文献中该方法也常与前文所述光谱学方法联用,用于分析激活剂离子的掺杂格位。 2019年,夏志国等[10]报道了Rb3YSi2O7∶Eu2+荧光粉中对Eu2+离子进行选择性格位占据以获得蓝光激发下的宽带红光发射(λex=450 nm,λem,max=622 nm)。在Rb3YSi2O7晶体结构(空间群P63/mmc)中存在形成[Rb1O9〗、[Rb2O6〗和[YO6〗三种配位多面体的阳离子格位可供Eu离子占据。利用TOPAS软件尝试通过结构精修确定Eu离子的占据格位。精修初始模型中设置Eu可同时占据上述三种阳离子格位,但精修结果表明只有Y3+格位有Eu离子占据,而其他阳离子格位Eu的占位率为零,见表1。但作者指出上述精修结果不能排除少量Eu离子占据Rb+格位的可能性:如表1所示,Eu离子占位率精修结果的标准偏差为0.000 8(0.08%),因此Rietveld精修不能给出Eu占位率低于0.24%(3倍标准偏差,即0.24%)的占位率精修结果[24]。荧光光谱结果表明该荧光粉表现出基于Eu2+离子4f→5d跃迁的宽带激发与发射特征。如表2所示,根据不同阳离子在不同配位数时的有效离子半径对比推测,占据Y3+离子格位的Eu离子应为Eu3+,而占据Rb+离子(如果有)的Eu离子应为Eu2+。Eu-L3边X射线吸收近边结构谱结果表明,该样品中所含Eu离子价态同时表现为3+(占主要)和2+(占次要)。进一步采用密度泛函理论计算方法探究Eu2+的占据格位,表明Eu2+倾向于多数占据Rb(2)+格位,少量占据Y3+格位。 表1 Rb3YSi2O7∶0.02Eu样品精修后所得原子坐标、占位率与各向同性位移参数[10] 表2 不同配位数时Eu2+、Eu3+、Rb+和Y3+离子的有效离子半径[10] 在高浓度掺杂的情况下,能否通过Rietveld精修方式确定激活剂离子的占据格位和占位率呢?梁宏斌等[13]报道了Ca6BaP4O17∶Eu2+荧光粉,随着Eu掺杂量的增加,其XRD主衍射峰逐渐向高角度偏移。收集Ca6-xBaEuxP4O17(x=0.01,0.05,0.11)样品的高质量XRD衍射图谱,采用Rietveld结构精修方法确定Eu2+离子在Ca2+/Ba2+格位的占位率,结果如图10和表3所示。根据精修所得结果,掺杂后的荧光粉的化学式应写为Ca6-3xBa1-2xEu5xP4O17。 表3 精修所得Ca6-xBaEuxP4O17(x=0,0.01,0.05,0.11)样品的化学式[13] 图10 (a)Ca6-xBaEuxP4O17的XRD图谱及在2θ=32.9°~33.4°区间的放大图;Ca6-xBaEuxP4O17的Rietveld精修结果:(b)x=0.01,(c)x=0.05,(d)x=0.11[13]。 上述结果对比表明,Rietveld结构精修在表征较低掺杂浓度掺杂离子的占据格位及占位率方面具有一定的局限性,在表征一系列从低掺杂浓度到高掺杂浓度样品中激活剂离子占据格位与占位率方面具有可参考性。 2.2.2 扩展X射线吸收谱技术 扩展X射线吸收谱技术是另一种可用以分析激活剂离子配位结构的手段。物质对X射线的吸收系数与入射X射线的能量有关,在一种原子吸收边(吸收系数突变处,内层电子被激发到外层而引起)高能侧几百电子伏范围内,吸收系数呈振荡变化。离吸收边30~50 eV直到近1 000 eV范围内的振荡被称为扩展X射线吸收精细结构(EXAFS),其形状与近邻原子结构状态即近邻配位原子种类、原子间距、配位数、无序度因子等有关,因而通过分析EXAFS谱可获得原子近邻结构信息[25]。 Pawik等[26]报道采用X射线吸收近边结构谱来证明不同压力条件下所合成SrSi2O2N2∶Eu2+荧光粉中Eu2+已完全还原为+2价,并采用EXAFS谱研究Eu2+在基质中的格位占据,发现Eu2+优先占据Sr2格位(占60%左右),剩余Eu2+占据Sr4格位。Akai等[27]报道采用EXAFS谱研究Ce3+在Ca3Sc2Si3O12中的格位占据,在Ca3Sc2Si3O12中存在八配位的Ca2+和六配位的Sc3+两种格位可供Ce3+占据。但如图11所示,在k=26~107 nm-1范围进行傅里叶转换后可见,Ce—O的峰值位置与Ca—O的一致,而与Sc—O的差异明显,因此推断Ce3+占据Ca2+格位。此外,Zhu等[28]通过EXAFS谱证明了在Mg14Ge5O24∶Mn4+荧光粉中引入F-和Ti4+离子后增强了其对蓝光的吸收效率的原因在于Mn—F键的形成。因此,EXAFS手段不仅可以确定激活剂离子的格位占据情况,还能判断其占位率;其对激活剂离子格位占据和占位率的判断(尤其是低浓度掺杂时)的精确度要远高于前述晶体结构精修手段。 图11 Ca3Sc2Si3O12∶Ce中Sc、Ca和Ce的K边EXAFS谱的傅里叶转换[27]。 采用理论计算如从头计算等第一原理计算方法分析Ce3+/Eu2+激活荧光粉中掺杂格位时,将从头计算所得到激活剂离子在不同格位占据时的4f→5d跃迁能量与荧光粉在低温下实测所得激发光谱进行对比,可确定Ce3+/Eu2+在荧光粉中的占据格位;另外,不同占据构型计算所得的形成能也用于辅助判断格位占据。相比于Ce3+掺杂复杂体系,Eu2+掺杂复杂体系的从头计算更加困难,因为Eu2+离子激发态4f65d1所涉及的能级数量远大于Ce3+离子激发态4f05d1所涉及的能级数量[29]。 图12 SrAl2O4中Eu2+占据Sr1和Sr2格位时其4f→5d跃迁(从4f7(8S7/2)基态到4f65d1电子组态的自旋八重态能级)的计算能量位置和相对振子强度示意图,同时给出归一化实测激发光谱用于对比[30]。 图13 Sr3AlO4F中和配位中心的4f1→5di(i =1~5)能级跃迁的计算能量位置和相对振子强度示意图,同时给出实测激发光谱用于对比[16]。 图14 (a)Sr4Al14O25∶0.1%Mn4+荧光粉的激发(黑线)与发射(红线)光谱,其中理论计算所得Mn4+的能级位置用竖线给出(长竖线,自旋四重态;短竖线,自旋二重态);(b)在Sr4Al14O25晶体结构中Mn4+倾向于占据Al4和Al5格位而不倾向于占据Al6格位示意图[33]。 白光LED用荧光粉中激活剂离子如Ce3+、Eu2+和Mn4+在基质中时常出现多格位占据现象,如何表征其多格位占据问题对于分析相应荧光粉的发光特性具有重要意义。本文综述了8种研究占据格位的方法,列于表4。这8种方法可归为三类:光谱学方法、结构分析法和计算光谱学方法。其中,光谱学方法包括以下5种谱图的测试分析:激发波长依赖的发光光谱与监测波长依赖的激发光谱、波长依赖的荧光衰减曲线、时间分辨发射光谱、变温发射光谱与变温荧光衰减曲线以及掺杂浓度依赖的发射光谱。 表4 各种激活剂离子格位占据(和/或占位率)分析方法的特点及优劣势比较 首先,上述方法在区分激活剂离子所占格位数方面各有优劣势。比如对不同延迟时间时测得的时间分辨发射光谱进行高斯分峰拟合,可区分激活剂离子在不同格位的发射能量并证明占据不同格位的激活剂离子间存在能量传递;而从掺杂浓度依赖的发射光谱中可以推断激活剂离子的优先占据格位,但需要制备一系列不同掺杂浓度的样品且激活剂离子在多个格位中的占据倾向性一致时,无法根据浓度猝灭现象区分占据单一格位还是多个格位。此外,当激活剂离子与基质被取代格位离子属于不等价取代时,尤其要区分激活剂离子是占据了不同晶体学格位还是占据同一格位但形成了多种微观配位多面体。 其次,值得强调的是,合成纯相样品是分析荧光粉中激活剂离子格位占据的前提。在荧光粉合成过程中,有时即使采取了多种方法对制备过程进行优化却依然难以去除杂相(如硼酸盐体系中非常容易产生稀土硼酸盐LnBO3杂相且难以去除[34];其他类似情况还有硅酸盐体系中易产生Ln2Si2O7∶Ce和/或Ln2SiO5∶Ce杂相,碱土硅酸盐体系中易产生(Ba,Sr,Ca)2SiO4∶Eu/Ce,以及磷酸盐体系中易产生稀土磷酸盐LnPO4∶Ce等)。尽管杂相的含量很少,但其发光却可能很强,若未加认真辨识分析,将会对激活剂离子在主相中的格位占据判断产生非常大的误导。 第三,激活剂离子掺杂进入基质后出现或选择性地仅占据某一格位、或占据多个格位但占据优先性显著不同、或几乎均匀地占据多个格位的这种现象的驱动力,很可能来自掺杂离子与基质中被取代格位的(平均)离子半径间的差异。梁宏斌等[34]报道,在未掺杂Ca3La3(BO3)5基质中,Ca2+和La3+是有序分布的,但当掺杂Ce3+后,Ce3+的离子半径介于Ca2+和La3+之间,其同时占据Ca2+和La3+两个阳离子格位但优先占据La3+格位,因为其与La3+离子的离子半径更加接近;此外,Ce3+占据Ca2+格位时还造成一定程度的反占位缺陷(即少量Ca2+占据La3+/Ce3+格位),用于电荷平衡补偿。而在其关于Ba1.995-xSrxSiO4∶0.005Eu2+中Eu2+格位占据分析的论文中报道[31],在x≤1时,Eu2+仅占据(平均)离子半径较小的AE2格位;当1 最后,在实际研究中,采用多种方法联用对准确分析激活剂离子的(优先)占据格位具有很大优势。比如,将计算光谱学方法与实验光谱学方法结合,可以推断出对应于实测激发光谱的具体微观配位多面体。再如,将结构分析方法与多种光谱学方法联用,不仅可以确定激活剂离子所占格位数,还能确定其优先占据格位及半定量占位率。此外,如表4中所述,EXAFS手段在表征激活剂离子格位占据及占位率方面有独特优势,相信随着我国同步辐射光源的建设,将会催生更多相关研究。 本文专家审稿意见及作者回复内容的下载地址:http://cjl.lightpublishing.cn/thesisDetails#10.37188/CJL.20210309.2 激活剂离子格位占据研究方法
2.1 光谱学方法






2.2 结构分析法





2.3 计算光谱学方法
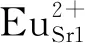




3 总结与展望
 猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31农业工程学报(2022年8期)2022-08-08贵州大学学报(自然科学版)(2022年2期)2022-03-31黑龙江大学自然科学学报(2022年1期)2022-03-29阅读(科学探秘)(2021年8期)2021-09-01科技资讯(2017年23期)2017-09-09饮食科学(2017年5期)2017-05-20中国民族民间医药·下半月(2016年12期)2017-01-19
猜你喜欢
北京航空航天大学学报(2022年8期)2022-08-31农业工程学报(2022年8期)2022-08-08贵州大学学报(自然科学版)(2022年2期)2022-03-31黑龙江大学自然科学学报(2022年1期)2022-03-29阅读(科学探秘)(2021年8期)2021-09-01科技资讯(2017年23期)2017-09-09饮食科学(2017年5期)2017-05-20中国民族民间医药·下半月(2016年12期)2017-01-19
