
基于第一性原理的锂空气电池钌掺杂石墨烯正极氧还原反应机理研究
2022-03-04 02:32张唐虎张添昱王金朋
原子与分子物理学报 2022年4期
张唐虎,李 强,张添昱,王金朋,孙 红
( 沈阳建筑大学 机械工程学院,沈阳 110168)
1 引 言
面对日益增长的能源危机和环境污染等挑战,具有高能量密度[1]和环境友好性[2]的新型储能电池—锂空气电池被广泛研究.其中,双电解液锂空气电池一方面由于每个氧分子的四电子转移具有最高的理论能量密度[3],另一方面可以避免锂枝晶以及过氧化锂电池综合性能的影响.双电解液锂空气电池主要由锂负极、有机电解液、锂离子导通膜、水系电解液以及空气正极组成,而正极一侧的催化材料往往决定了电池性能的优异程度.多年来,众多的科研人员为进一步提高电池性能从事研究正极催化剂,众多催化材料中性能较好的一直是铂和铂基催化剂[4-8],但铂催化剂的高成本和稀有性等问题使其无法大规模应用于锂空气电池.因此,为了克服这些问题,人们付出了巨大的努力来寻找非铂甚至非金属的催化剂[9,10].
催化剂的选择,需考虑到双电解液锂空气电池正极要拥有足够的氧气以及产物容纳场所,因此催化电极材料需要具备较高电导率和比表面积以提供高效反应的场所.自2004 年石墨烯的发现以来,因其复合材料具有高比表面积、优良的电导率和优异的化学稳定性在锂空气电池领域备受关注[11].为探索石墨烯掺杂电子特性,杂原子掺杂的石墨烯得到了深入研究,当石墨烯中引入杂原子后,其原有的电子性质会发生明显的改变,因此整体的化学活性会有所提高[12].Zhang 等人通过密度泛函理论方法将磷( P) 原子替换石墨烯中的碳原子,研究表明在一定程度上提高了反应速度和降低了反应能垒[13].对于其他一些非金属原子,如硼( B) 、硫( S) 等原子掺杂石墨烯同样被Feng 等人证明具有较高的催化性能[14].对于金属原子,Varghese、Hu 等人通过DFT 方法证实了镓( Ga) 、铁( Fe) 和钴( Co) 等杂原子掺杂石墨烯同样具有较好的氧还原反应( Oxygen Reduction Reaction,ORR) 催化性能[15,16].同样,催化剂应还具备高的催化效率,Jiang、Bai 等人通过实验发现Ru 基催化剂具有较高的氧还原催化效率[17,18].对于过渡金属Ru 的研究,Nandi 等人利用第一性原理研究了Ru 纳米团簇的催化效率,结果表明Ru在ORR 反应中能够与中间体良好的结合,表现出了良好的催化性能[19].鉴于Ru 掺杂石墨烯材料具有较高的导电率、比表面积和催化效率,适合成为双电解液锂空气电池催化电极,但对于它的ORR 催化机理尚不明确,由此须进一步研究Ru掺杂单层石墨烯在双电解液锂空气电池中的催化氧还原性能以及相应的氧还原反应机理.
本文主要采用的是第一性原理研究方法,使用Materials Studio 软件的Dmol3程序包计算n( n=1 ~3) Ru 原子掺杂单层石墨烯的结构性质以及对O2分子的吸附性能,揭示其在碱性条件中的催化氧还原反应机理.研究了Ru 原子掺杂单层石墨烯对催化活性影响的机理,对研制新型ORR 催化剂有着重要的指导意义.
2 计算方法和模型
2.1 计算方法
本文所进行的模拟计算都是基于密度泛函理论( Density Function Theory,DFT) 的第一性原理Dmol3程序,自旋不受到限制.广义梯度近似( Generalized Gradient Approximation,GGA) 下 的Perdew-Burke-Ernzerhof( PBE) 泛函[20]被用作电子间交换关联函数.基组为双数值轨道基组+p轨道极化函数DNP,总轨道截止选择的数值为5.1 Å,采用全电子相对论( All Electron Relativistic) 处理金属原子的相对论效应.布里渊区积分采用3 ×3 ×1 的K 点进行结构优化.原子结构优化中的能量收敛性判据为1.0e -6 Ha,Hellmann-Feynman 原子力收敛判据为每个原子受力不大于0.002 Ha/Å,最大位移为0.005 Å.通过使用Complete LST/QST 方法搜索过渡态,确保频率分析时只存在一个虚频.
定义吸附能如下式:

其中,Eadsorb表示吸附能,Eslab和Eadsorbate分别代表隔离的掺钌石墨烯和氧气的能量,Eslab-adsorbate表示掺钌石墨烯片吸附氧气的总能量.所有原子结构的能量都是在完全弛豫后计算的,吸附能计算值为负值时表明发生放热反应,Eadsorb越负表示吸附能越大,吸附体系越稳定.
2.2 计算模型
采用Materials Studio 软件构建石墨烯晶胞模型并对其进行结构优化,图1( a) 为结构优化后的石墨烯结构示意图,其碳碳键长与晶格长度分别为1.420 Å 和3.21 Å,这与以前的计算结果基本一致[21].将优化后的石墨烯截取(001) 面并建立4×4 ×1 的单层石墨烯超晶胞,其中包含了32 个C原子.计算中,取X 和Y 方向在单层石墨烯平面内,Z 方向垂直于单层石墨烯平面,沿Z 方向设置了20 Å 的真空层避免层与层之间的相互作用.
3 结果与讨论
基于建立的Ru 掺杂单层石墨烯表面模型,完成对Ru 原子掺杂单层石墨烯的稳定性以及对O2分子的还原能力的研究,对经掺杂的单层石墨烯表面氧还原反应机理进行详细研究.最终通过评估反应能垒以及反应产物OH 基团的吸附能大小,确定材料对于ORR 反应的催化性能.
3.1 Ru 掺杂单层石墨烯的几何结构和电子性质
3.1.1 Ru 掺杂单层石墨烯的几何结构
首先我们将n( n=1 ~3) 个Ru 原子依次替换掉石墨烯中的C 原子,经过充分驰豫后获得最稳定结构,图1 中红圈位置为Ru 掺杂位点.图1 为原始单层石墨烯以及nRu( n=1 ~3) 掺杂的单层石墨烯的俯视图,分别将其命名为Gr、Ru1-Gr、Ru2- Gr、Ru3- Gr.从优化的结构可以看出,nRu 掺杂的单层石墨烯保留了原始单层石墨烯的平面结构,但由于Ru 原子半径较大,经掺杂后原始的碳碳键的键长发生了变化.为进一步证明nRu( n=1 ~3) 原子掺杂单层石墨烯结构的可靠性,对其表面进行了动力学模拟.模拟结果表明在500 K 温度下、5 ps 的时间内,表面的掺杂原子依然位于掺杂位点.
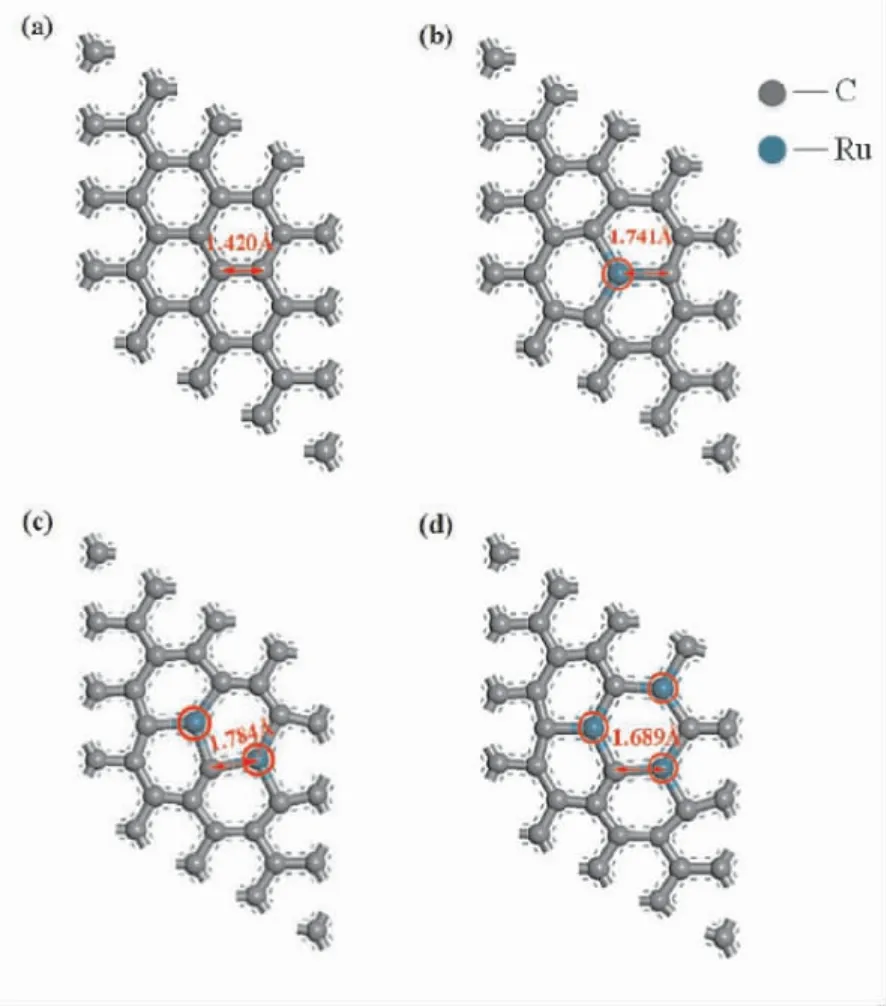
图1 单层石墨烯及Run -Gr 充分弛豫后的结构: (a)Gr; (b)Ru1 -Gr; (c)Ru2 -Gr; (d)Ru3 -GrFig.1 Fully relaxed structures of the monolayer graphene and Run -Gr: ( a) Gr; ( b) Ru1 -Gr;( c) Ru2 -Gr; ( d) Ru3 -Gr
3.1.2 Ru 掺杂石墨烯的电子性质
下图为Gr、Ru1-Gr、Ru2-Gr 以及Ru3-Gr的能带结构.从图2( a) 中可以看出,原始单层石墨烯是直接零带隙半导体,其导带和价带在狄拉克点处重合,与其他文献报道出的计算结果相吻合[22].从图2( b) 中可以看出当单Ru 进行掺杂石墨烯时,使得费米能级移至价带,可判断掺杂形式为p 形掺杂,进一步提高了整体的导电性.随着掺杂原子数的增加,锥点会进一步打开,杂质浓度的增加导致体系内聚能的降低,表明键的弱化,电子性质会变的更加灵活.
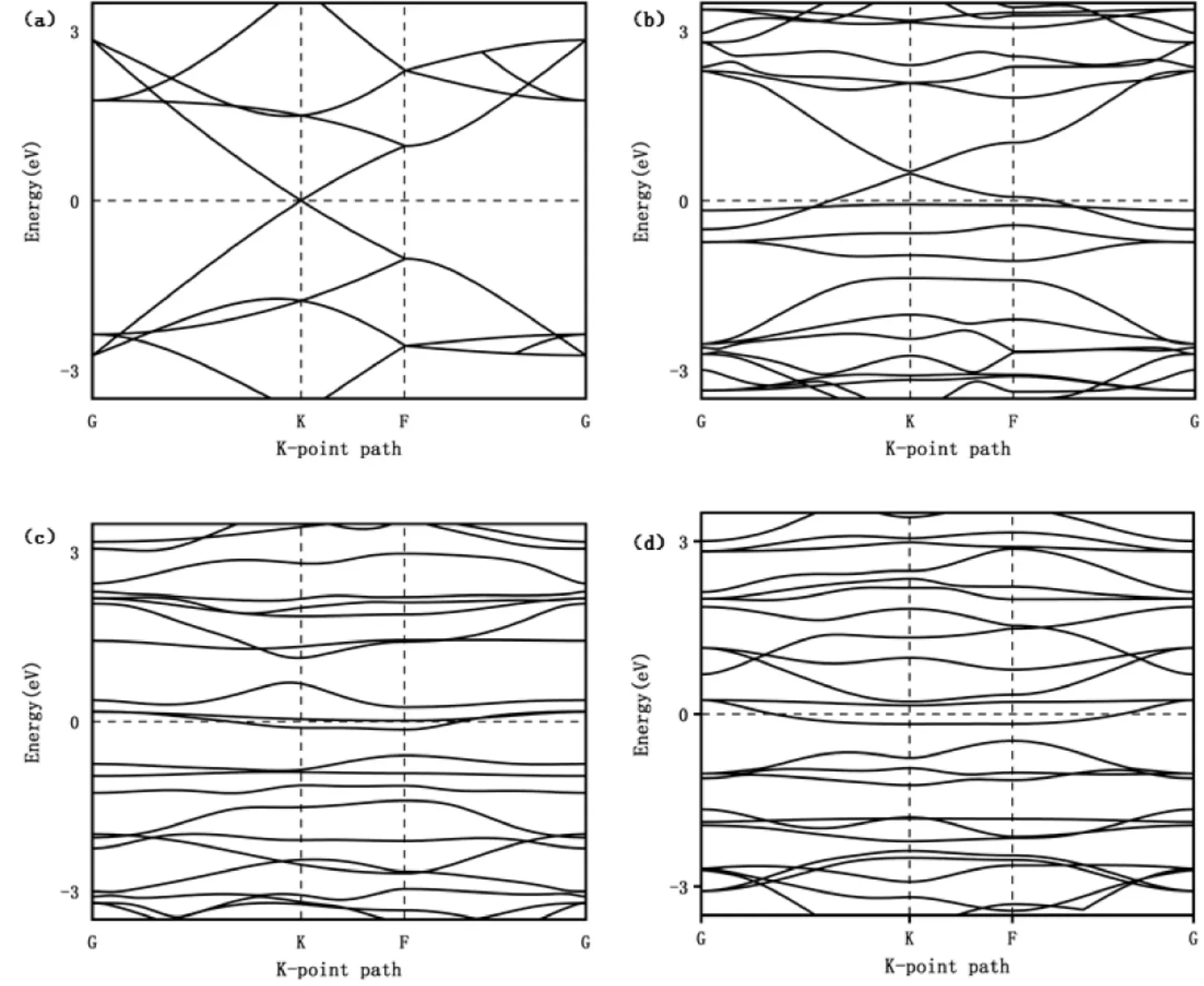
图2 不同掺杂体系的能带结构: ( a) Gr; ( b) Ru1 -Gr; ( c) Ru2 -Gr; ( d) Ru3 -GrFig.2 Band structure of different doping systems: ( a) Gr; ( b) Ru1 -Gr; ( c) Ru2 -Gr; ( d) Ru3 -Gr
3.2 O2分子在Ru 掺杂单层石墨烯表面的吸附
空气正极侧发生的氧还原反应是以O2分子吸附为开端.原始单层石墨烯表面性质并不活泼,O2分子物理吸附在石墨烯表面,吸附能为-0.26 eV,并不利于后续氧还原反应的进行.针对三种掺杂结构对于O2的吸附情况,计算不同的吸附位点的吸附能,确定最稳定的能量吸附构型.根据吸附状态可计算得到O2吸附能、O2键长、Ru-O 键长以及密立根( Mulliken) 电荷布居数,如下表中.从表1 中,可以发现,随着Ru 原子掺杂数量的增加,对于O2分子的吸附作用越强,两个O 原子之间的距离也会变的更加远; 当掺杂原子数量由1 增加至3 时,三种掺杂体系中Ru 原子获得的电子也会增加,尤其O2在Ru3-Gr 表面吸附时,两个O 原子的吸附状态并不相同,出现了两个Ru-O 键长数值.
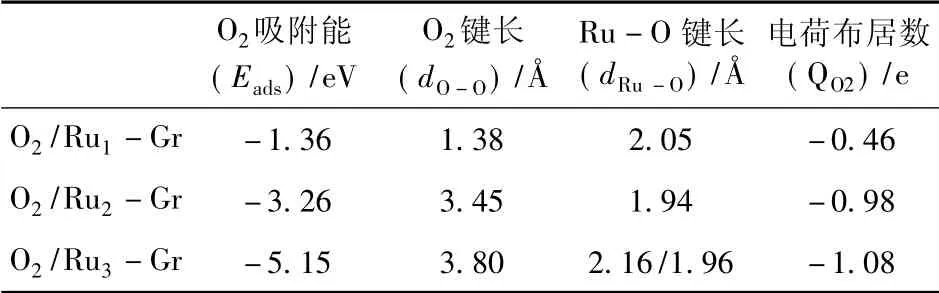
表1 不同Ru 掺杂结构吸附O2分子的性质Table 1 Adsorption properties of O2 adsorbed on the Ru1 -Gr、Ru2 -Gr and Ru3 -Gr
图4 为不同掺杂结构吸附O2分子的最稳定构型.如图4( a) Ru1-Gr 所示,O2分子稳定吸附在Ru 原子顶位,此时对应的Ru-O 键长为2.05 Å,O2的吸附能为-1.36 eV,大于Gr 对O2的吸附能,同时O2失去了0.46 e 的电荷.经Ru 掺杂后会引起石墨烯的变形,O2分子被吸附后,会进一步增加局部的形变,从而增加系统的局部反应性[23].如图4( b) 、( c) ,分别为O2分子在Ru2-Gr 以及Ru3-Gr 表面的吸附.随着掺杂Ru 原子数量的增加,反应表面可以提供更多O2的吸附点位,同时Ru 原子之间距离较远,所以O2分子在发生吸附时,分子键会发生断裂,不需要跨过能量壁垒,O 原子会直接与Ru 原子成键.因此可以看出,当掺杂数量变多时,O2分子在对应的表面会直接解离吸附,使得后续的反应会更加顺利的进行.
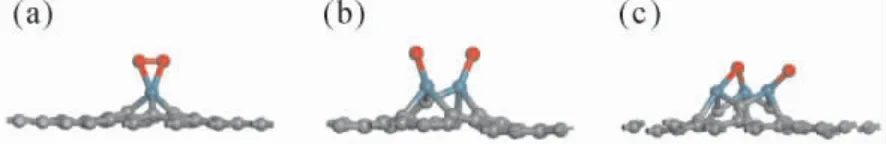
图4 优化后的O2 分子在不同表面的吸附构型:( a) Ru1 -Gr; ( b) Ru2 -Gr; ( c) Ru3 -GrFig.4 Optimized adsorption configurations of O2 adsorbed on the Run -Gr: ( a) Ru1 - Gr; ( b)Ru2 -Gr; ( c) Ru3 -Gr
本文为进一步了解O2分子的吸附机理,通过计算其态密度分析其电子性质.图5 给出了三Ru掺杂单层石墨烯中Ru 原子的4d 轨道及吸附的O2分子的PDOS 图.在图中可以看到吸附后的O2分子与Ru 原子的4d 电子轨道发生了重叠,产生了较强的杂化,说明Ru 原子和吸附的氧原子之间形成了化学键.上述研究表明Ru3-Gr 表面对O2分子有较好的吸附及还原效果.
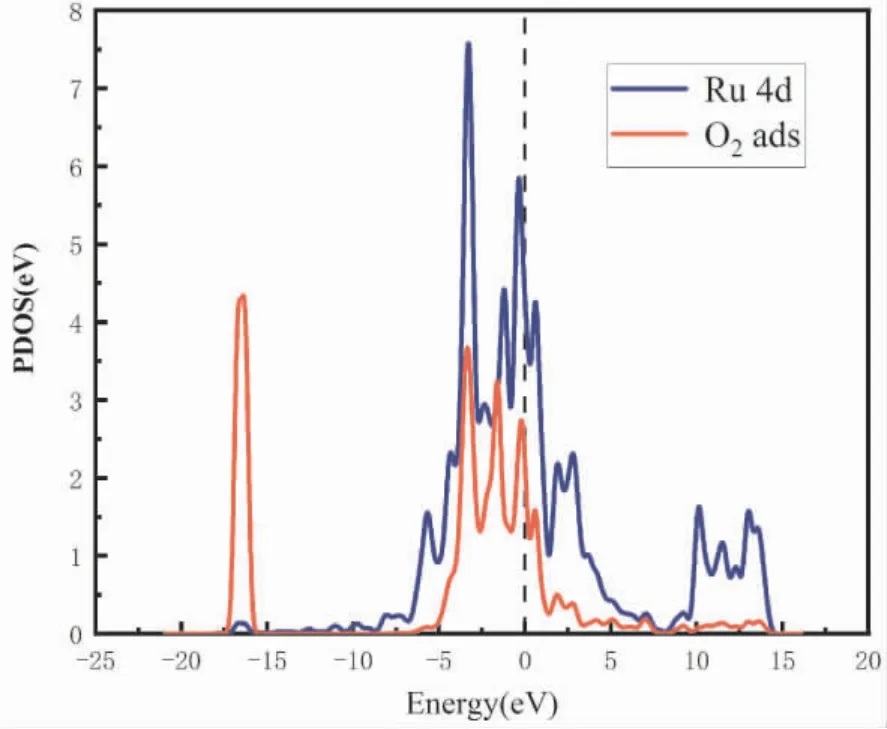
图5 Ru3 -Gr 分波态密度( PDOS)Fig.5 O2 adsorbed Ru3 - Gr surface’s Partial density of states ( PDOS)
3.3 氧还原反应机理
在碱性环境中,溶液中只有氢氧根离子,吸附的O2分子首先与周围的水分子进行反应,形成四个OH 基团,形成的OH 基团会得到由电极提供的电子,进入溶液中形成游离的OH.所以,在碱性环境中总反应是生成OH-的4e-路径,ORR总反应公式为: O2+2H2O+4e-→4OH-.
3.3.1 Ru1-Gr 表面的氧还原反应
作为ORR 的开始,首先考虑O2的吸附.通过将O2分子放在Ru1-Gr 的不同位置并比较它们的吸附能,我们确定了最有利的能量吸附构型,如图6 所示.
经多个位点的测试,在Ru1-Gr 表面吸附的O2分子未直接解离,且没有得到O2解离相应的过渡态.吸附的O2分子会与周边的水分子直接反应.
图6( a) 为Ru1-Gr 表面氧还原反应机理图,在IS 中,O2分子吸附在Ru 原子上后,占据了吸附位点.当首个H2O 分子加入后,在自然条件下,氧气分子之间的双键会断开,形成两个O*.同时水分子中的一个H 原子解离向其中一个O*移动形成OH* ,自身变成一个OH* ,整个过程是一个自然反应,不需要跨过反应能垒,此时的能量变化为-0.98 eV.
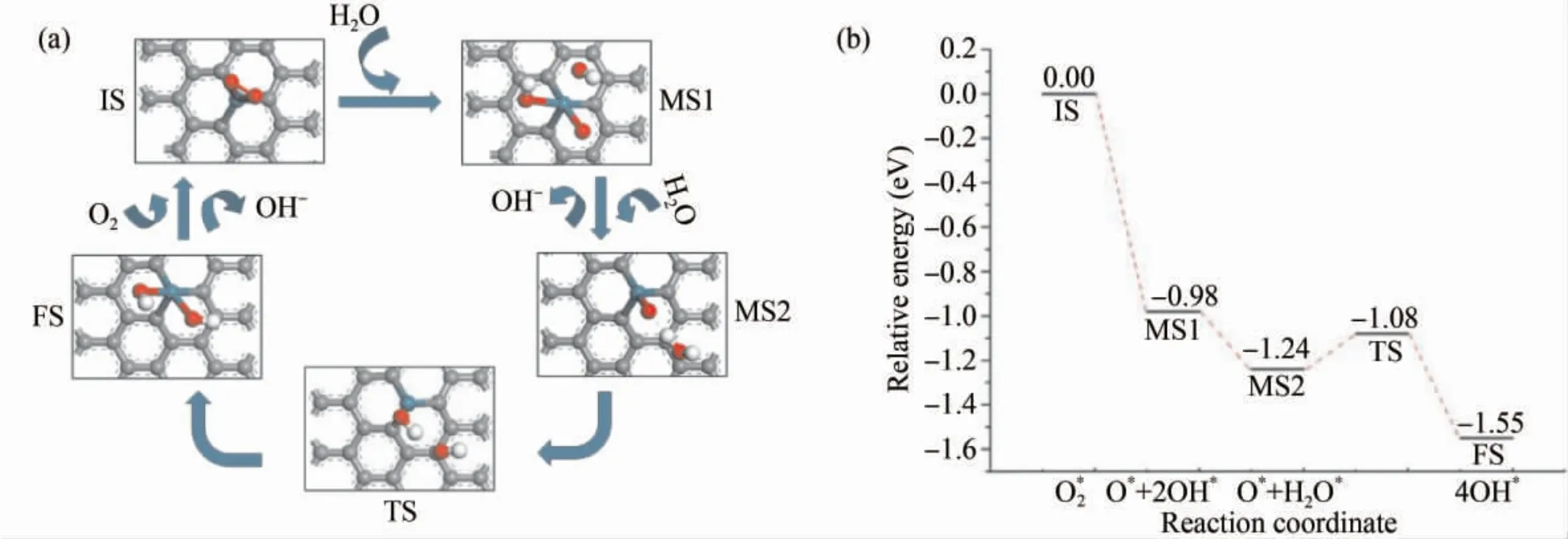
图6 碱性条件下Ru1 -Gr 表面( a) 氧还原反应的初态IS、中间态MS、过渡态TS 及终态FS 的稳定结构( b) 相应的反应能量路径Fig.6 ( a) The optimized structure configuration of initial state( IS) ,intermediated state( MS) ,transition state( TS) ,and final state( FS) of ORR on Ru1 -Gr in alkaline environment; ( b) The corresponding reaction energy paths
在OH* 得到外电路提供的电子后,会以游离状态的OH-脱去吸附,剩余的一个O 原子依然是吸附在Ru 原子之上.同理,当第二个H2O 分子继续参加反应时,因吸附位点的缺少,所以依旧只能通过氢键的形式吸附于Ru -O 键斜上方,此时H2O 分子吸附的能量变化为-0.26 eV.反应再次生成两个OH* ,同时吸附在Ru 原子上,该步反应需要跨过0.16 eV 的能垒,反应的能量变化为-0.31 eV,属于放热反应,这表明该步反应在热力学是稳定的,释放的能量有利于OH 基团的脱离.在Ru1-Gr 表面进行的ORR 反应中,能垒的存在使得状态MS2 到终态FS 成为整个反应的限制性步骤.考虑到后续反应的进行,碱性条件下氧还原反应产物OH* 在Ru1-Gr 表面的吸附能经计算为-8.38 eV,可以发现此时的吸附状态非常稳定,不易发生OH* 的脱吸附反应.
根据O2分子在Ru1-Gr 基底表面的吸附以及解离情况,ORR 的分步反应公式如下,式中* 代表吸附物种:
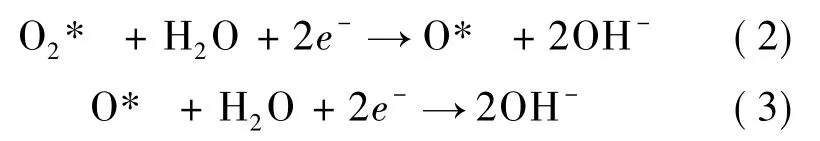
3.3.2 Ru2-Gr 表面的氧还原反应
同Ru1- Gr 表面类似,通过将O2分子放在Ru2-Gr 的不同位置并确定了最有利的能量吸附构型,如图7 所示.
图7( a) 描述了Ru2-Gr 表面催化氧还原反应的可能反应途径以及各反应步骤的最稳定结构.由于活性位点的增加,O2分子会得到直接的解离.在反应分子的吸附模型MS1 中,H2O 分子通过氢键吸附于解离后的O* ,H2O 分子吸附的能量变化为- 0.94 eV.然后通过克服一个约为0.05 eV的能量壁垒后,H2O 分子中的一个H 原子会得到解离,与O* 结合生成OH*.对应于过渡态TS 中可以看到,一个H 原子会先与Ru 原子上的O 原子成键生成OH* ,留下的OH 基团会继续向Ru 原子方向移动,最终吸附在基底表面的Ru 原子上.在MS2 中,该步反应最终生成两个OH 基团共同吸附在相同的一个Ru 原子上,该步反应的能量变化为-1.92 eV,属于放热反应,说明了产物有很好的稳定性.
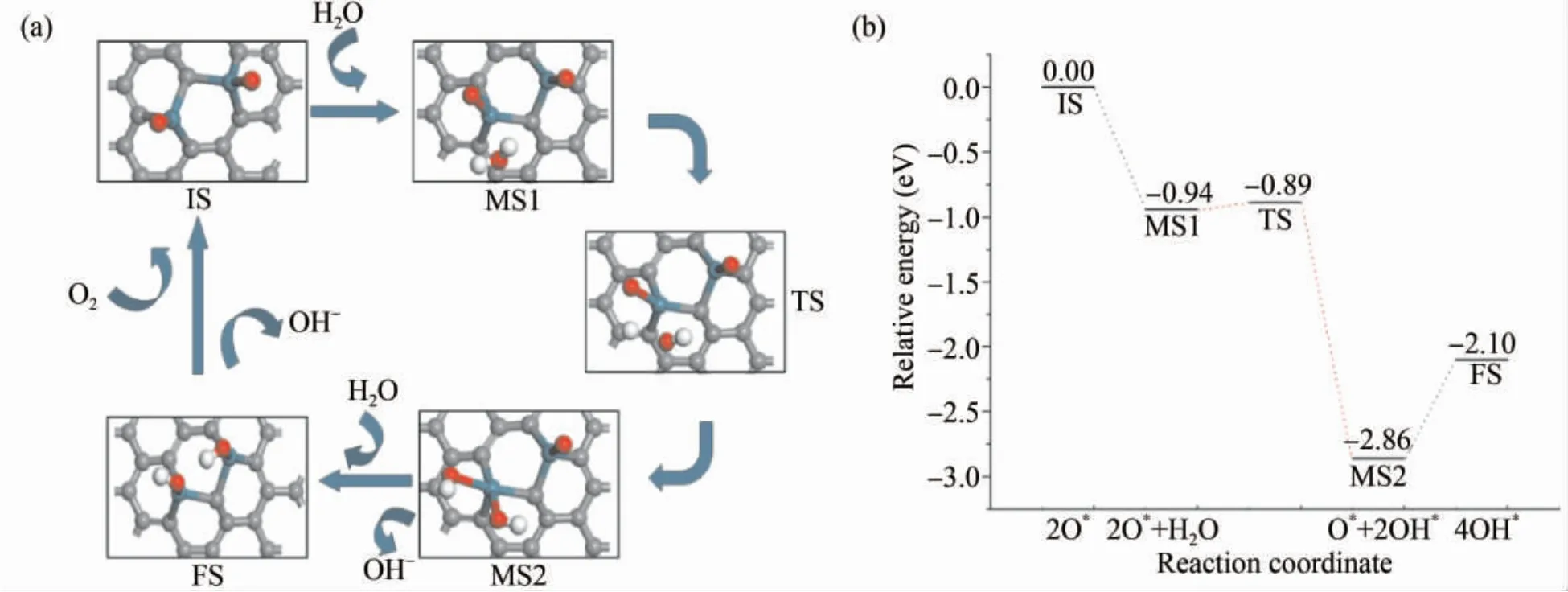
图7 碱性条件下Ru2 -Gr 表面( a) 氧还原反应的初态IS、中间态MS、过渡态TS 及终态FS 的稳定结构( b) 相应反应能量路径Fig.7 ( a) The optimized structure configuration of initial state( IS) ,intermediated state( MS) ,transition state( TS) ,and final state( FS) of ORR on Ru2 -Gr in alkaline environment; ( b) The corresponding reaction energy paths
在OH* 脱吸附后,第二个水分子会继续参加反应.因为一方面O 原子具有较强的电负性,另一方面表面暴露出Ru 这一活性点位,H2O 分子会吸附在暴露的Ru 原子处,同时H2O 分子的一个H 原子直接解离,与O* 成键.该步反应最终生成两个OH* ,从反应路径中可以看出此过程没有生成过渡态,但从状态MS2 到状态FS 需要进行吸热,不利于ORR 的进行,成为Ru2-Gr表面ORR 反应中的主要限制性步骤.氧还原反应产物OH* 在Ru2-Gr 表面的吸附能为-5.19 eV,相较于Ru1-Gr,OH* 的吸附能减小,使反应变的容易进行.
根据O2分子在Ru2-Gr 基底表面的吸附以及解离情况,ORR 的分步反应公式如下:
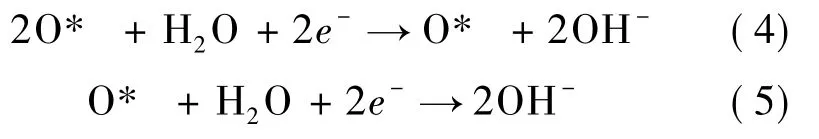
3.3.3 Ru3-Gr 表面的氧还原反应
在Ru3-Gr 催化剂表面,因为O2分子会直接解离成两个O* ,所以表面的氧还原反应步骤基本与Ru2-Gr 表面一致.图8( a) 为Ru3-Gr 表面的氧还原反应过程,随着活性点位的进一步增加,在该表面的氧还原反应无需任何的能量壁垒.如图8( a) 中的MS1 所示,系统中加入H2O 分子后,H2O 分子中的H 原子会直接解离并与O* 形成OH* ,剩余一个OH 基团会继续吸附在暴露的Ru 原子上,这一步反应生成两个OH 基团,该步反应的能量变话为-4.99 eV.随后第二个H2O 分子与另一个O 原子反应继续生成两个OH 基团.同样可计算OH* 在Ru3- Gr 表面的吸附能为-3.27 eV,吸附能进一步减小.通过对比可以看出,三Ru 掺杂单层石墨烯具有更好的催化作用.
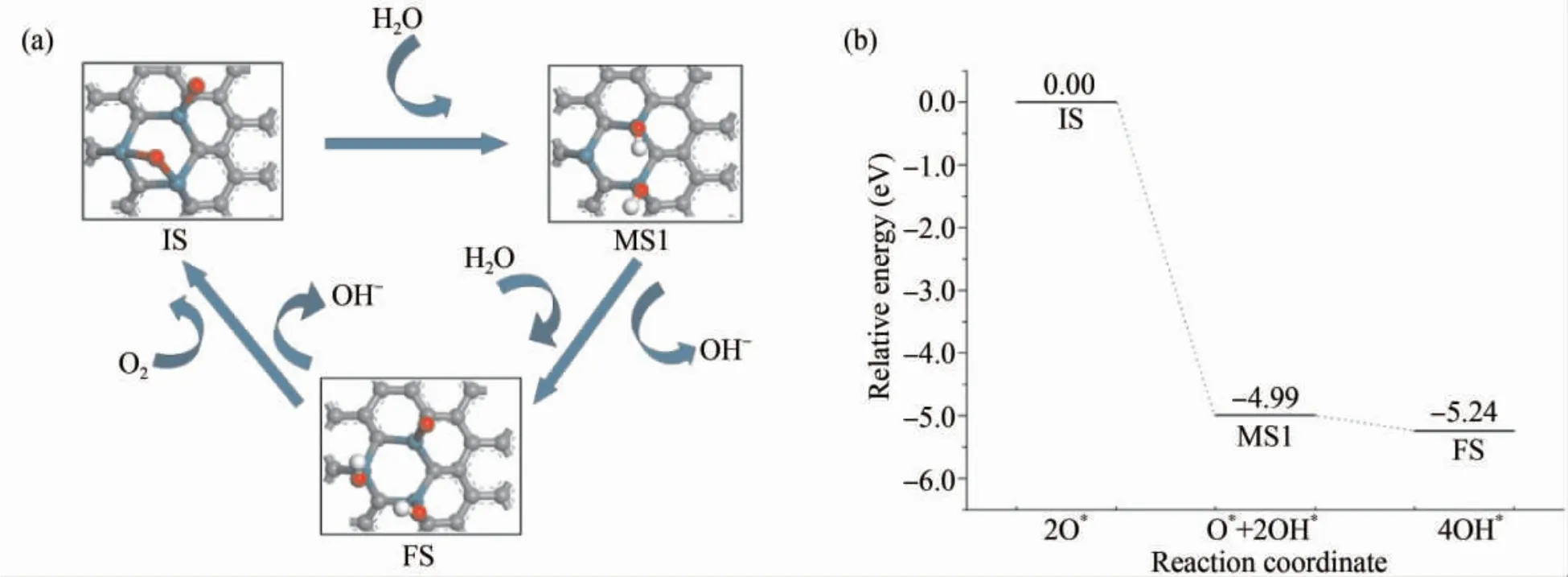
图8 碱性条件下Ru3 -Gr 表面( a) 氧还原反应的初态IS、中间态MS、过渡态TS 及终态FS 的稳定结构( b) 相应反应能量路径Fig.8 ( a) The optimized structure configuration of initial state( IS) ,intermediated state( MS) ,transition state( TS) ,and final state( FS) of ORR on Ru3 -Gr in alkaline environment; ( b) The corresponding reaction energy paths
根据O2分子在Ru3-Gr 基底表面的吸附以及解离情况,ORR 的分步反应公式如下:
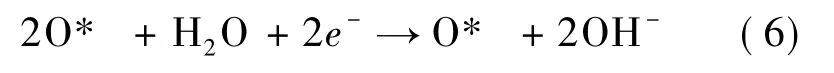
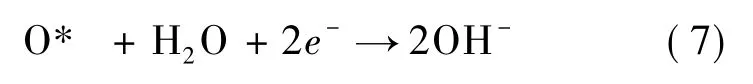
对比不同掺杂体系氧还原反应的过程,在Ru1-Gr 中会出现一个中间过渡态,其对应的反应能垒为0.16 eV.在Ru2-Gr 中同样出现了中间过渡态,其对应的反应能垒为0.05 eV,相比Ru1-Gr 体系,能量壁垒发生了减小,使得反应能更加容易发生.当在Ru3-Gr 表面发生氧还原反应时,并没有过渡态及相应能量壁垒的产生,整个过程都是在自然状态中发生.同时考虑到碱性条件下产物OH 基团的影响,它在Gr、Ru1- Gr、Ru2-Gr 及Ru3-Gr 表面的吸附能分别为-0.76 eV、-8.38 eV、-5.19 eV 和-3.27 eV.因为原始单层石墨烯表面性质不活泼,所以它对于OH基团的吸附作用较低,当单Ru 进行掺杂后,吸附能增大,不利于OH 基团的脱吸附,而随着掺杂数量的增加,OH 基团在Ru2- Gr 及Ru3- Gr表面的吸附能相较于Ru1-Gr 表面又逐渐减小,而吸附能的减小使得氧还原反应更加容易进行.
4 结 论
本文基于密度泛函理论,探究了n( n=1 ~3)Ru 原子掺杂单层石墨烯在碱性条件下的ORR 反应催化机理,得到如下结论:
(1) 相比较O2分子在原始单层石墨烯的吸附能( -0.26 eV) ,O2分子在Ru1-Gr 表面的吸附能提高到了-1.36 eV,随着掺杂原子数量的增加,O2在Ru2- Gr 和Ru3- Gr 吸附能分别为-3.26 eV、-5.15 eV,由此可见三Ru 掺杂石墨烯与O2形成了更为稳定的吸附结构.
(2) 单Ru 掺杂会使得原始石墨烯中的狄拉克点上移,双Ru 及三Ru 掺杂会进一步使得狄拉克点打开,从而使得Ru 掺杂石墨烯的导电性能显著提升.
(3) 随着Ru 原子在石墨烯中掺杂数目的逐渐增加,反应能垒由0.16 eV 降低至0.05 eV,而OH 基团吸附能由-8.38 eV、 -5.19 eV 变化至-3.27 eV,研究表明三Ru 原子掺杂的单层石墨烯有利于碱性环境下的ORR 反应进行,反应过程中无过渡态产生,表明三Ru 掺杂石墨烯可作为潜在的ORR 催化剂应用于双电解液锂空气电池领域.
猜你喜欢
分子催化(2022年1期)2022-11-02
北京航空航天大学学报(2022年5期)2022-06-06
新课程·下旬(2019年7期)2019-09-17
电子制作(2019年15期)2019-08-27
表面工程与再制造(2019年6期)2019-08-24
铜仁学院学报(2018年6期)2018-07-05
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
