
186 例矮小症患者遗传学检测结果分析
2022-05-16 04:54王莉莉吴海瑛谢蓉蓉王凤云陈秀丽王晓艳张丹丹陈临琪
临床儿科杂志 2022年5期
王莉莉 吴海瑛 谢蓉蓉 王凤云 陈 婷 陈秀丽 孙 辉 王晓艳 张丹丹 陈临琪
苏州大学附属儿童医院内分泌遗传代谢科(江苏苏州 215000)
矮小症是最常见的儿童内分泌遗传代谢疾病之一,其发病率约为3.0%~5.0%[1-2]。生长发育是一个复杂的动态过程,受遗传、环境、营养、疾病及内分泌激素等多种因素的共同影响。其中遗传因素约占80.0%,是成人终身高的主要决定因素[3]。由于临床表型和遗传异质性,潜在的病因往往难以发现。随着基因检测技术包括拷贝数变异分析、全外显子测序等在矮小病因研究中的应用,发现一些单基因变异、染色体片段缺失或重复是导致儿童矮小症的遗传病因[4]。但是,由于矮小症遗传学研究还非常有限,且基因检测费用较高,尚未在临床普遍应用。本研究的主旨为①明确哪些矮小症患者需要进行基因检测,为矮小症儿童基因检测的临床应用提供依据;②明确矮小症的相关基因,为其精准诊疗提供基础。
1 对象与方法
1.1 研究对象
选取2017年1月—2020年10月因生长缓慢至苏州大学附属儿童医院内分泌遗传代谢科就诊,未明确病因的186例矮小症患儿。矮小症诊断标准:身高低于同性别、同年龄、同种族正常健康儿童平均身高2个标准差以上,或低于第三百分位数(参照首都儿科研究所生长发育研究室制定的《0~18岁儿童青少年身高、体质量标准差单位数值表》)[2]。
纳入标准:年龄<16岁,符合矮小症诊断标准,伴有下列1项或以上表现,①矮小家族史,患儿父母及其家族中有1人或多人身材矮小(男性<160 cm,女性<150 cm);②特殊面容,包括前额突出、眼距宽、眼睑下垂、睑裂长、睑裂下斜、睑裂上斜、斜视、蓝巩膜、鼻梁低平、高腭弓、小下颌、面中部发育不良、嘴唇厚、耳位低、招风耳、颈短、颈蹼;③骨骼发育异常,包括四肢短、双下肢弯曲、鸡胸、漏斗胸、手指短、手指弯曲、多指(趾)、并指(趾)、脊柱畸形、马德隆畸形、关节过度屈曲、髌骨小或缺如、骨质异常、Chiari畸形;④先天性心脏发育异常,包括房间隔缺损、室间隔缺损、法洛四联征、卵圆孔未闭、主动脉狭窄、肺动脉狭窄、动脉导管未闭、肺动脉高压;⑤其他系统异常,包括语言发育落后、运动发育落后、智力低下、泌尿生殖器畸形、听力障碍(听力下降、先天性感音性耳聋)、皮肤咖啡斑、多毛;⑥严重矮小,即身高低于同年龄、同性别儿童平均身高-3 SD 或低于父母遗传靶身高的-3 SD;⑦无生长追赶的小于胎龄儿(SGA)。
排除标准:①颅内肿瘤、占位,颅脑外伤或放射损伤;②甲状腺功能减退症(甲状腺发育不良、桥本甲状腺炎);③Turner综合征;④21三体综合征;⑤其他染色体数目异常;⑥慢性系统性疾病,如慢性肾脏病、血液系统疾病、肿瘤、长期应用糖皮质激素。
本研究获苏州大学附属儿童医院伦理委员会批准(批准文号:202003)及家长知情同意。
1.2 方法
1.2.1 临床资料收集 包括矮小症患儿的身高、年龄、性别、临床表型、矮小家族史等资料。
1.2.2 全外显子基因测序和基因芯片检测 入院后采集患儿及其父母外周血各2 mL,应用全血磁珠纯化试剂盒提取DNA,利用芯片IDTxGen Exome Research Panel v 1.0 对进行捕获,采用 Illumina Novaseq 6000 平台测序,测序数据经Illumina Sequence Control Software(SCS)评估合格后,进行数据读取和生物信息学分析。原始测序数据去除污染和接头序列,然后利用BWA软件(http://bio-bwa.sourceforge.net/)将过滤后的序列比对到NCBI数据库人类基因组参考序列(hg 19)上,利用GATK 软件(https://software.broadinstitute.org/gatk/)分析得出单核苷酸变异(single nucleotide variation,SNV)和插入缺失突变(inserts and deletions,INDEL)的相关信息。然后通过ANNOVAR 软件(http://annovar.openbioinformatics.org/en/latest/)对所有的SNP 和INDEL 进行注释。单核苷酸变异利用SIFT(http://sift.jcvi.org/),PolyPhen-2(http://genetics.bwh.harvard.edu/pph2/),Mutation Taster(http://www.mutationtaster.org/)软件进行致病性预测和保守性预测。根据美国医学遗传学和基因组学学院(ACMG)的最新标准和指南对候选变异进行分类。经过分析筛选得到的获选变异位点利用PCR和Sanger测序验证,并在家系成员中进行共分离验证。PCR引物对利用Primer 5.0设计。PCR产物经过Sanger测序并在ABI 3730XL DNA 测序仪(Applied Biosystems,Thermo Fisher Scientific,USA)上进行分析。对于全外显子基因测序发现的可能的染色体片段拷贝数变异进一步完善基因芯片检查,明确儿童矮小症病因诊断,根据患儿临床表型及有无矮小家族史进行分组,比较不同分组间基因检测的阳性率。
1.3 统计学分析
采用SPSS 25.0 统计软件进行数据分析。应用GraphPad软件进行绘图。非正态分布的计量资料以中位数(P25~P75)表示。计数资料以例数(百分比)表示,组间比较采用χ2检验。采用二元logistic回归模型分析矮小症患儿基因检测阳性的预测因素。以P<0.05为差异有统计学意义。
2 结果
2.1 一般临床资料
共纳入186 例矮小症患儿,中位年龄为7.3(5.1~9.1)岁,男103例、女83例。有矮小家族史85例(45.7%),伴特殊面容52例(28.0%),伴骨骼系统异常51例(27.4%),SGA 26例(14.0%),伴心血管系统异常25例(13.4%),伴泌尿生殖系统畸形23例(12.4%),伴智力低下7例(3.8%),伴皮肤毛发异常5例(2.7%),伴听力障碍3例(1.6%)。
2.2 基因检测结果
本研究共发现6 9 例阳性结果,总检出率37.1%,单基因变异54例(表1),染色体片段缺失15例(包括DiGeorge综合征4例,Williams综合征3例,Leri-Weill软骨发育不全2例,16p11.2微缺失综合征2例、Xp22.33-p22.31缺失、17p13.3缺失、15q26.3缺失、2q31.1缺失各1例)。

表1 单基因变异54例致病机制分类
2.3 基因阳性和阴性组之间临床表型比较
根据基因检测结果分为阳性组和阴性组。阳性组身高标准差<-3 SD、伴骨骼发育异常、伴特殊面容、伴先天性心血管发育异常、伴>1个异常表型的比例高于阴性组,矮小家族史的比例低于阴性组,差异均有统计学意义(P<0.05)。见表2。

表2 基因检测阳性组与阴性组临床表型比较[n (%)]
将85 例有矮小家族史的患儿根据是否伴有异常表型及按矮小程度分组进行比较发现,不同矮小程度组间基因检测阳性率差异有统计学意义(P<0.05),身高标准差<-4 SD 组的阳性率较高;有矮小家族史且伴异常表型患儿的基因检测阳性率高于仅有矮小家族史患儿,差异有统计学意义(P<0.05)。见表3。
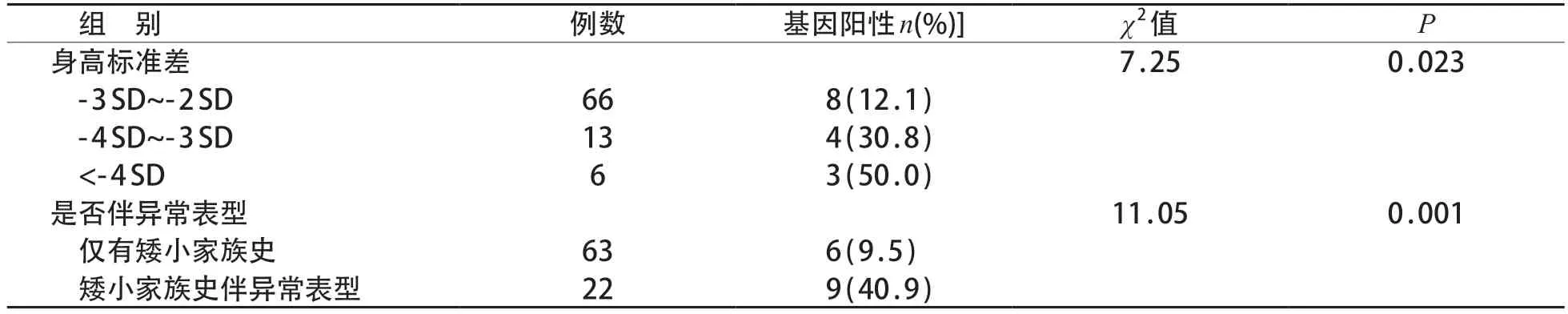
表3 85例有矮小家族史伴或不伴其他表型及不同矮小程度组间阳性率比较
2.4 不明原因矮小症患儿基因检测阳性发生的预测因素
将单因素分析中差异有统计学意义的因素纳入二元logistic回归分析,结果见表4,特殊面容和骨骼发育异常是儿童矮小症基因检测结果阳性发生的预测因素(P均<0.05)。
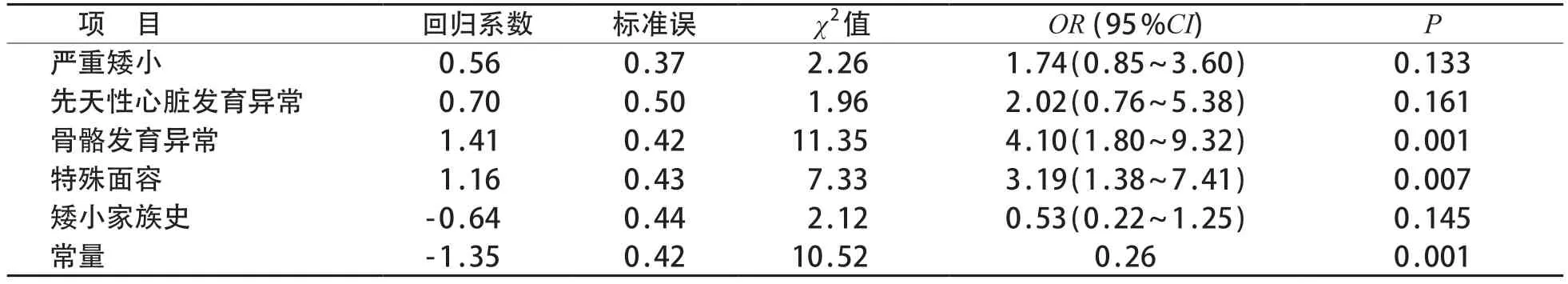
表4 不明原因矮小症患儿基因检测阳性发生的预测因素
3 讨论
身材矮小是患者及其家属非常关注的问题,也是儿童内分泌科常见的疾病之一。遗传因素对身高变异的影响很大程度上归因于许多不同基因的综合作用,这意味着身高通常可能是一个多基因遗传的复杂性状[5-6]。然而,严重身材矮小往往是由单基因变异引起的,从而对身高产生较大的影响(如FGFR3、NPR2、ACAN等)[7]。单基因变异导致的疾病复杂多样,导致的矮小可能是匀称的或是非匀称的、严重的或轻微的、可以发生在出生前和/或出生后[8]。此外,一个基因的不同变异类型可以导致不同的临床表型谱,一种临床表型可能是由不同的基因变异引起[9]。一些基因的变异不仅会损害骨骼生长板的发育和/或功能,还会损害非骨骼系统,导致综合征性的矮小[10]。
近年来,遗传学方法包括基因组测序技术和生物信息学方法,尤其是二代测序技术(next generation sequenxcing,NGS),已经成为鉴定单基因矮小遗传病因的重要工具[11-13]。过去下丘脑垂体-生长激素(GH)-类胰岛素样生长因子-1(IGF-1)轴被认为是儿童生长发育的主要调控途径[14],然而最近研究发现许多新的基因变异会导致身材矮小,其中绝大多数不参与GH-IGF-1 系统。因此有研究认为下丘脑垂体-GH-IGF-1 轴只是控制生长板软骨生长的众多调控系统之一,儿童的正常生长不仅需要正常浓度的GH 和IGF-1,还需要多种其他激素、旁分泌因子,以及软骨细胞增殖、肥大和细胞外基质产生所需的多种细胞内信号通路共同作用的调控[12,15]。根据对骨骼生长板作用的机制不同进行分类,包括内分泌生长激素轴信号通路、旁分泌信号、软骨细胞外基质和细胞内信号通路的遗传缺陷[16]。调控这些信号通路上的基因变异均可导致身材矮小。本研究发现2 例GH 1基因变异,均表现为严重矮小、具有生长激素缺乏的典型面容(前额突出,眼距宽,鼻梁低平),且GH1基因复合杂合变异较单杂合变异的矮小程度更严重,GH激发试验及IGF-1检测对于诊断有一定的帮助,明确诊断仍需借助基因测序。文献报道ACAN基因变异的患儿多有骨龄超前,伴有骨骼发育异常[17],本研究中5例ACAN基因变异患儿中,仅1例存在骨龄超前,家系中携带者及先证者均无骨骼发育异常,提示ACAN基因变异临床表型谱广泛,虽无早发性骨关节炎或骨骼畸形、骨龄超前的患儿仍不能完全排除ACAN基因变异的可能。FGFR 3基因杂合变异可导致软骨发育不全,临床特征为不成比例的身材矮小,四肢短小,头大、前额突出、面中部发育不良,“三叉手”、关节松弛,腰椎后凸畸形等[18]。本研究发现1例FGFR3基因变异(c.1663G>T,p.V555L)患儿仅表现为身材矮小,身材比例正常,无明显骨骼畸形,母亲身高148 cm,身材匀称。因此建议对匀称性身材矮小且有矮小家族史的患儿,仍需要考虑FGFR3基因变异。本研究中发现2 例ANKRD 11基因变异导致的KBG 综合征,均为尚未报道的位点,患儿均有典型的上颌中切牙过大、身材矮小、小指弯曲、轻中度智力落后,均无心脏发育异常。另外本研究中的男性患儿还有隐睾、异位肾的表现,有文献报道部分KBG综合征男性患儿可有隐睾表现[19]。但尚无异位肾的报道,具体机制有待进一步研究。
本研究在186 例矮小患儿中共检出69 例阳性结果,仅2例为GH-IGF-1轴相关基因,23例为骨骼生长板相关基因,44 例为综合征性相关基因。提示内分泌GH 轴异常导致的矮小症可能相对更罕见。因此建议临床医师除了关注GH 轴的异常,应该更多重视骨骼发育系统以及其他系统的异常。2014年Dauber等[20]建议对所有身高低于-3SD的个体,或身高低于-2.5 SD 且具有一种或多种临床表型(包括多发性垂体激素缺乏症表型、明确的GH不敏感、SGA缺乏追赶成长、骨骼发育不良、智力障碍、小头畸形及其他先天性异常或畸形),或身高低于父母遗传靶身高-2 SD 的患儿均应进行基因检测,这些患儿可能存在单基因变异的风险,二代测序技术有助于明确其遗传病因。2018年国内研究者对114例中国矮小患儿依次进行靶基因/全外显子组测序和染色体微阵列分析,其中41 例明确了遗传病因,阳性率为36%。其中伴有面部畸形或骨骼异常患儿的阳性率显著高于无相应表型的患儿[21]。除上述研究外,国内外的此类研究并不多,目前国内对矮小症的诊治大多停留在传统的检测方法如GH 激发检测、IGF-1 的测定等,但传统的检测方法仅能明确1.0%~40.0%矮小症的病因[20,22],仍有大部分患儿病因尚未明确。随着遗传学基因检测技术的快速发展,希望可以有更多身材矮小的患儿得到明确的病因诊断,从而达到精准治疗。本研究对186例病因不明的身材矮小患儿进行全外显子测序,发现了69例阳性结果,其中54例为单基因变异,15例为基因芯片证实的染色体片段缺失变异,总检出率为37.1%。
本研究共有85例患儿具有矮小家族史,其中仅有矮小家族史63例,有矮小家族史且伴有其他异常表型22例,共检出15例阳性结果。有矮小家族史且伴有其他异常表型组的阳性率高于仅有矮小家族史不伴异常表型组,推测仅有矮小家族史的患者更多的可能是多基因遗传[23]。当合并有其他异常表型时,提示患者存在单基因变异的可能性较大,建议进行基因检测明确病因。本研究结果提示特殊面容、骨骼发育异常是儿童矮小症基因检测阳性发生的预测因素。这与相关研究结果一致[21]。因此对于以矮小就诊且合并有特殊面容和/或骨骼发育异常的患儿,应建议其完善基因检测,进一步明确矮小病因。
综上所述,在病因不明的矮小症患儿中存在潜在基因变异的可能,全外显子测序技术是检测儿童矮小症遗传病因的有效技术手段。儿童矮小症的遗传病因主要涉及GH 轴相关基因、骨骼生长板相关基因及矮小综合征相关基因。具有特殊面容和/或骨骼发育异常患儿的诊断率明显高于无相应表型的患儿,提示这两种表型可作为矮小患儿基因检测阳性发生的预测因素,但需要收集更多的样本来进一步验证。
猜你喜欢
作物学报(2022年2期)2022-11-06
中国现代医生(2022年21期)2022-08-22
肝博士(2022年3期)2022-06-30
农村科学实验(2022年2期)2022-03-12
中国听力语言康复科学杂志(2021年6期)2021-12-21
支部建设(2020年15期)2020-07-08
三农资讯半月报(2020年2期)2020-03-09
风湿病与关节炎(2018年1期)2018-02-11
华文文学(2017年2期)2017-05-07
百科知识(2015年18期)2015-09-10
