
Genomic Epidemiology of lmported Cases of COVlD-19 in Guangdong Province,China,October 2020–May 2021*
2022-06-10 10:35LIANGDanWANGTaoLIJiaoJiaoGUANDaWeiZHANGGuanTingLIANGYuFengLIAnAnHONGWenShanWANGLiCHENMengLinDENGXiaoLingCHENFengJuanPANXingFeiJIAHongLingLEIChunLiangandKEChangWen2
LIANG Dan ,WANG Tao ,LI Jiao Jiao,GUAN Da Wei,ZHANG Guan Ting,LIANG Yu Feng ,LI An An,HONG Wen Shan,WANG Li,CHEN Meng Lin,DENG Xiao Ling,CHEN Feng Juan,PAN Xing Fei,JIA Hong Ling,LEI Chun Liang,#,and KE Chang Wen2,,,,,#
1.Guangdong Provincial Key Laboratory of Virology,Institute of Medical Microbiology,Jinan University,Guangzhou 510632,Guangdong,China;2.Guangzhou National Laboratory,Guangzhou 510700,Guangdong,China;3.Guangdong Provincial Center for disease control and prevention,Guangdong Workstation for Emerging Infectious Disease Control and Prevention,Guangzhou 511430,Guangdong,China;4.Shantou University Medical College,Shantou 515041,Guangdong,China;5.Guangzhou Eighth People’s Hospital,Guangzhou 510030,Guangdong,China;6.School of public health,Sun Yat-sen University,Guangzhou 510080,Guangdong,China;7.School of Public Health,Southern Medical University,Guangzhou 510515,Guangdong,China;8.Guangzhou Mendel Genomics and Medical Technology Co.,Ltd.,Guangzhou 510535,Guangdong,China;9.Department of Infectious Diseases,the Third Affiliated Hospital of Guangzhou Medical University,Guangzhou 510150,Guangdong,China;10.Department of Medical Biochemistry and Molecular Biology,School of Medicine,Jinan University,Guangzhou 510632,Guangdong,China
Abstract Objective The pandemic of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) has been engendering enormous hazards to the world.We obtained the complete genome sequences of SARSCoV-2 from imported cases admitted to the Guangzhou Eighth People’s Hospital,which was appointed by the Guangdong provincial government to treat coronavirus disease 2019 (COVID-19).The SARS-CoV-2 diversity was analyzed,and the mutation characteristics,time,and regional trend of variant emergence were evaluated.Methods In total,177 throat swab samples were obtained from COVID-19 patients (from October 2020 to May 2021).High-throughput sequencing technology was used to detect the viral sequences of patients infected with SARS-CoV-2.Phylogenetic and molecular evolutionary analyses were used to evaluate the mutation characteristics and the time and regional trends of variants.Results We observed that the imported cases mainly occurred after January 2021,peaking in May 2021,with the highest proportion observed from cases originating from the United States.The main lineages were found in Europe,Africa,and North America,and B.1.1.7 and B.1.351 were the two major sublineages.Sublineage B.1.618 was the Asian lineage (Indian) found in this study,and B.1.1.228 was not included in the lineage list of the Pangolin web.A reasonably high homology was observed among all samples.The total frequency of mutations showed that the open reading frame 1a (ORF1a) protein had the highest mutation density at the nucleotide level,and the D614G mutation in the spike protein was the commonest at the amino acid level.Most importantly,we identified some amino acid mutations in positions S,ORF7b,and ORF9b,and they have neither been reported on the Global Initiative of Sharing All Influenza Data nor published in PubMed among all missense mutations.Conclusion These results suggested the diversity of lineages and sublineages and the high homology at the amino acid level among imported cases infected with SARS-CoV-2 in Guangdong Province,China.
Key words:Severe acute respiratory syndrome coronavirus 2; Next-Generation sequencing;Phylogenetic tree;Variants
INTRODUCTION
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2),the pathogenic agent of coronavirus disease 2019 (COVID-19),has caused a global pandemic and conferred a profound effect on all aspects of society.As of June 15,2021,175 million infections have been confirmed,and 3.8 million deaths have been reported all over the world[1]. The Chinese government managed to contain the virus through a series of draconian control measures.However,the number of newly confirmed cases outside of China has been rising rapidly.Thus,China prioritized protecting itself from imported cases from other countries or territories.
Guangdong Province is known as the“South Gate of China,”with a large number of foreign passengers;hence,the most number of imported cases was detected here compared with other regions of Mainland China[2].The World Health Organization announced several variants of concern(VOCs) and variants of interest (VOIs) of SARS-CoV-2 on May 31,2021[3].With the increase in the number of genome sequences available in the public database,analysis using more sequences by sampling from a wider area over a longer period was imperative.In the present study,we employed second-generation sequencing to obtain virus sequence information and performed advanced comprehensive methods to analyze the genetic characteristics of SARS-CoV-2 in the imported cases based on the demographic and baseline characteristics of these patients. Our studies enriched the knowledge of the existing epidemic strains of SARS-CoV-2 in the world and provided valuable clues for defense against this infectious disease imported to China.
MATERIALS AND METHODS
Ethics Approval Statement
The protocol of the study was reviewed and approved by Guangdong Provincial Center for Disease Control and Prevention (approval number:W96-027E-202121).
Sample Sources
This study included human throat swab samples that were collected and processed in the Guangdong Provincial Center for Disease Control and Prevention,China,and throat swab samples,which were deidentified prior to analysis,collected from the Guangzhou Eighth People’s Hospital from October 2020 to May 2021.All the samples were inactivated at 56 °C for 30 min.Demographic characteristics and clinical information were collected from electronic medical records.
Nucleic acid Test (NAT) and Next-generation Sequencing (NGS)
All the throat swab samples were screened by quantitative real-time polymerase chain reaction method using the SARS-CoV-2 NAT kits (BioGerm,Shanghai,China).The positive specimens were processed for NGS using the MGISEQ-2000 platform of the Beijing Genomics Institute (BGI,Shenzhen,China).DNA fragment assembly was performed using the iGenome®VirusDetector system (BGI,Shenzhen,China),with the Wuhan-hu-01 (GenBank access no.MN908947.3) genome as the reference sequence. All ambiguous nucleotides in the alignment analysis were set to N.The ratio of non-N greater than 85% indicated a good fragment assembly in accordance with the manufacturer’s instruction.
Phylogenetic Analysis
The complete reference genomes were retrieved from the National Center for Biotechnology Information database (https://www.ncbi.nlm.nih.gov/).Pangolin web tool (https://cov-lineages.org/)was used for the origin of clades.Mattf v7.037(Kazutaka Katoh,Japan,2002) and BioEdit v7.0.9.0(Borland,USA,2009) were applied to sequence alignment and trimming,respectively. The maximum-likelihood (ML) phylogenetic tree was constructed using IQ-Tree v1.6.12 (Bui Quang Minh,Nguyen Lam Tung,Olga Chernomor,et al.,USA,2011).A total of 1,000 bootstrap replicates were used to deduce branch support.The phylogenetic tree was visualized using FigTree v1.4.2 (Andrew Rambaut,UK,2006) and Adobe Illustrator CS6(Adobe,USA,1987).In addition,the Nextclade web tool (https://clades.nextstrain.org) was used for putting up information about nucleotide mutations and amino acid mutations.R v3.6.1 (Ross Ihaka,Robert Gentleman,New Zealand,1995) was used to calculate the genetic distance of sequences,obtain the homology,and cleanse the mutation data at the nucleotide and amino acid levels.
Statistical Analyses
The data were managed using EpiData v3.1 (Jens M.Lauritsen,Mark Myatt,Michael Bruus,DK,1999).R v3.6.1 (Ross Ihaka,Robert Gentleman,New Zealand,1995) was used for descriptive analysis and plotting.All the bar graphs were drawn using the GraphPad Prism software v8.0.2 (San Diego,CA,USA,1992).
Age variables were expressed using“median{Interquartile range [(IQR): 25%–75%]},”and categorical variables including age group,gender,and NAT results were presented as percentages.A two-tailedP-value of < 0.05 was considered statistically significant throughout the study.
RESULTS
Demographic Information of the Patients
A total of 177 patients were included in this study,and their demographic characteristics are shown in Table 1.More than half of the respondents were male (136,76.84%).A total of 78 individuals(44.07%) were aged 31–40 years,and 6 people were over 61.The minimum age was 16,and the maximum was 85 (median age,34 years).A total of 123 out of 177 (69.49%) individuals showed positive results for SARS-CoV-2 RNA by the NATs,and among them,117 cases (95.12%) were sequenced successfully.Exactly 43 patients in our studies(24.29%) were asymptomatic cases (Table 1).

Table 1.Demographic information of patients with SARS-CoV-2 infection
Sequence Distribution in the Imported Cases
A total of 117 complete genomes sequences were obtained from this study,and the time and regional distribution through sequence clades are shown in Figure 1.With the sampling dated between June 2020 and May 2021,the time distributions of 177 patients were mainly in May 2021 (65/177,36.72%),February 2021 (43/177,24.29%),January 2021 (33/177,18.64%),and March 2021 (24/177,13.56%) (Figure 1A),and the time distributions of sequences were primarily in May 2021 (40/117,34.19%),January 2020 (25/117,21.37%),February 2020 (24/117,20.51%),and March 2021 (18/117,15.38%). The sequence distribution relevant to the timelines wasasymmetrical due to the successful epidemic control in China after April 2020[4].The sequences from this study were classified into 39 sublineages:A,A.23.1,B.1,B.1.1,B.1.1.157,B.1.1.214,B.1.1.222,B.1.1.228,B.1.1.368,B.1.1.420,B.1.1.7,B.1.170,B.1.177.81,B.1.2,B.1.214.2,B.1.351,B.1.36,B.1.36.16,B.1.36.17,B.1.36.38,B.1.36.22,B.1.402,B.1.413,B.1.427,B.1.429,B.1.438.1,B.1.466.2,B.1.468,B.1.524,B.1.525,B.1.533,B.1.580,B.1.595,B.1.596,B.1.618,B.1.84,C.36,L.3,and P.2,where the clades located were chiefly concentrated on B.1.1.7 (28/117,23.93%) and B.1.351 (20/117,17.09%).The lineage distribution included Europe (n=50),Africa (n=28),North America (n=22),Asia (excluding China) (n=12),China (n=3),South Africa (n=1),South America(n=1),and B.1.1.228 (n=1) (Figure 1B).The largest diversity of lineages was observed in January 2021,with six lineages,followed by February 2021,with five lineages.European and African lineages mainly appeared in May 2021 (25/117,21.37%vs.10/117,8.55%),the North American lineage emerged in January 2021 (11/117,9.40%),and that from South America and variant B.1.1.228 materialized in January 2021 (not classified on the Pangolin web tool).China lineages were interspersed in October 2020 and January and February 2021 (Figure 1C).

Figure 1.Distribution of imported cases.(A)Time distribution of 177 imported cases during the whole study period (from June 2020 to May 2021);(B) total number of lineages(including those from Europe,Africa,North America,Asia (excluding China),China,and South America and sublineage B.1.1.228) in 117 imported cases;(C) temporal distribution of each lineage in 117 imported cases.
The regional distribution was chiefly imported from the United States (USA;13/117,11.11%),Zambia (8/117,6.84%),Nigeria (8/117,6.84%),and the United Arab Emirates (UAE;6/117,5.13%).Additionally,the regional distribution included Indonesia (n=5),Cambodia (n=5),Tanzania (n=4),Bangladesh (n=4).Notably,the UK strains of the European lineage (B.1.1.7 and B.1.36.2) mostly came from Cambodia and Nigeria.The USA strains of the North American lineage (B.1.1.222,B.1.1.368,B.1.2,B.1.402,B.1.427,B.1.429,B.1.580,B.1.595,and B.1.596) were predominantly from the USA.The two patients infected with sublineage B.1.618 (Indian lineage) were asymptomatic and imported from UAE and Mozambique.Sublineage B.1.170,belonging to the European lineage (Egypt and UAE),first appeared in the world on March 19,2020,but occurred in June 2020 in our study.Sublineage B.1.468 (Asian lineage (Indonesia),a member of the early epidemiological transmission chains,first emerged on February 13,2020,and was included in our study in July 2020.
Phylogenetic Tree and Variation Analyses of SARSCoV-2 at the Nucleotide and Amino Acids Levels
For this study,a GTR+F+I+G4 nucleotide substitution model,the best-fit model based on the Akaike Information Criterion according to the Bayesian information criterion,was used for the phylogenetic analyses.The virus species in this study clustered in two large groups.Most viral sequences from patients with SARS-CoV-2 infection clustered in the African,North American,and European lineages.The rest of the sublineages were scattered in diverse branches of the phylogenetic tree.The diversity of molecular evolution of SARS-CoV-2 cases imported to Guangdong Province was revealed,which was consistent with the case distribution (left part of Figure 3).In the homology analyses,a comparison of the reference sequences with the sequences of respective samples indicated that the homology among genomic sequences was 99.68%–100%.In addition,the viral genomes from infected patients were consistent with the animal reference sequences,including theCanis lupusfamily,feline,mink,ferret,Panthera leo,tiger,andNeovison vison(99.76%–99.98%). Moreover,we analyzed the correlations between age,gender,clinical classification,and VOCs,with a statistically significant difference between males and females (P<0.05,not shown in the table).
The mutations in nucleotide that occurred in 117 sequences were found in these loci:S (nt23403),1a(nt241,3037),1b (nt14408),3a (nt25563),N(nt28881–28883),8 (nt27972),E (nt26456),and M(nt26735).Position 1a (nt241 and nt3037) exhibited the highest mutation rate,with T substituting for C,followed by nt14408 (position 1b) with C displacing T. At the amino acid level,mutations were discovered in the following sites:S (aa614),1b(aa314),1a (aa3676–aa3677),N (aa203–aa205),3a(aa57),and 8 (aa52).D614G had the highest mutation rate,followed by P314L (Figure 2 and Table 2).Supplementary Table S1 (available in www.besjournal.com) lists all substitutions in the nucleotides and amino acids.
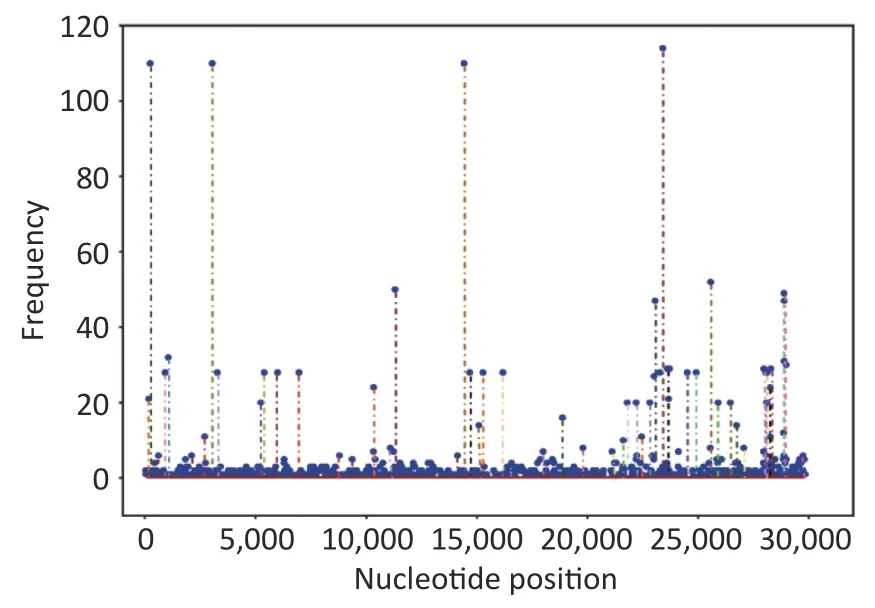
Figure 2.Frequencies of all mutations on the genome at the nucleotide level.
Supplementary Tables S2 and S3 (available in www.besjournal.com) show the nonsense and missense mutation distributions in the nonsynonymous mutation.Additionally,Supplementary Tables S4 and S5 (available in www.besjournal.com)list the reported amino acid mutation loci on the Global Initiative of Sharing All Influenza Data(GISAID) website (https://www.gisaid.org/) and publications in PubMed (https://pubmed.ncbi.nlm.nih.gov/),respectively.In total,1,076 nonsense mutations were observed on 476 nucleotide sites in all the analyzed sequences.A total of 2,010 missense mutations were present in 441 loci,and they were largely disseminated in positions ORF 1a and S.The missense mutations were demonstrated in all 12 gene fragments (right part of Figure 3).We also identified some missense mutations that had neither been reported on the GISAID database nor published in PubMed,including position non-S (replacements at five amino acid residue lineages,including those from the North American (sublineage B.1.2):Ile5Val of ORF9b and European lineages (sublineage B.1.214.2:Ala75Val of ORF9b;sublineage B.1.1.7:Met1-and Leu6Met of ORF7b;sublineage B.1.1.7:Met1-and Leu6Met of ORF7b and Arg25Ile of ORF9b),and position S (eight substitution sites,including those in lineages from North America(sublineage B.1.595):Leu226Phe;Europe (sublineage B.1): Tyr248Asn; North America (sublineage B.1.402):Cys1247Arg;Asia (not China) (B.1.618):Tyr145-and His146-;Asia (not China) (B.1.1.25):His66Asp; Africa (sublineage L.3): Gly75Arg,Tyr1155Phe;Africa (sublineage L.3):Tyr1155Phe,whose corresponding coding nucleotide sites were demonstrated as the points marked in red in the right panel of Figure 4.

Figure 3.Maximum-likelihood phylogenetic dendrogram constructed based on Pangolin’s classifications from the 117 sequences and Wuhan-hu-01 in GenBank as a reference sequence.The positions of all missense mutation sites on genes and their distribution,regardless of whether they were published or reported or unpublished and unreported,are revealed in the right part of the graph.

Figure 4.Maximum-likelihood phylogenetic dendrogram.The red dots in the right panel indicate that the missense mutations were not reported and not published.
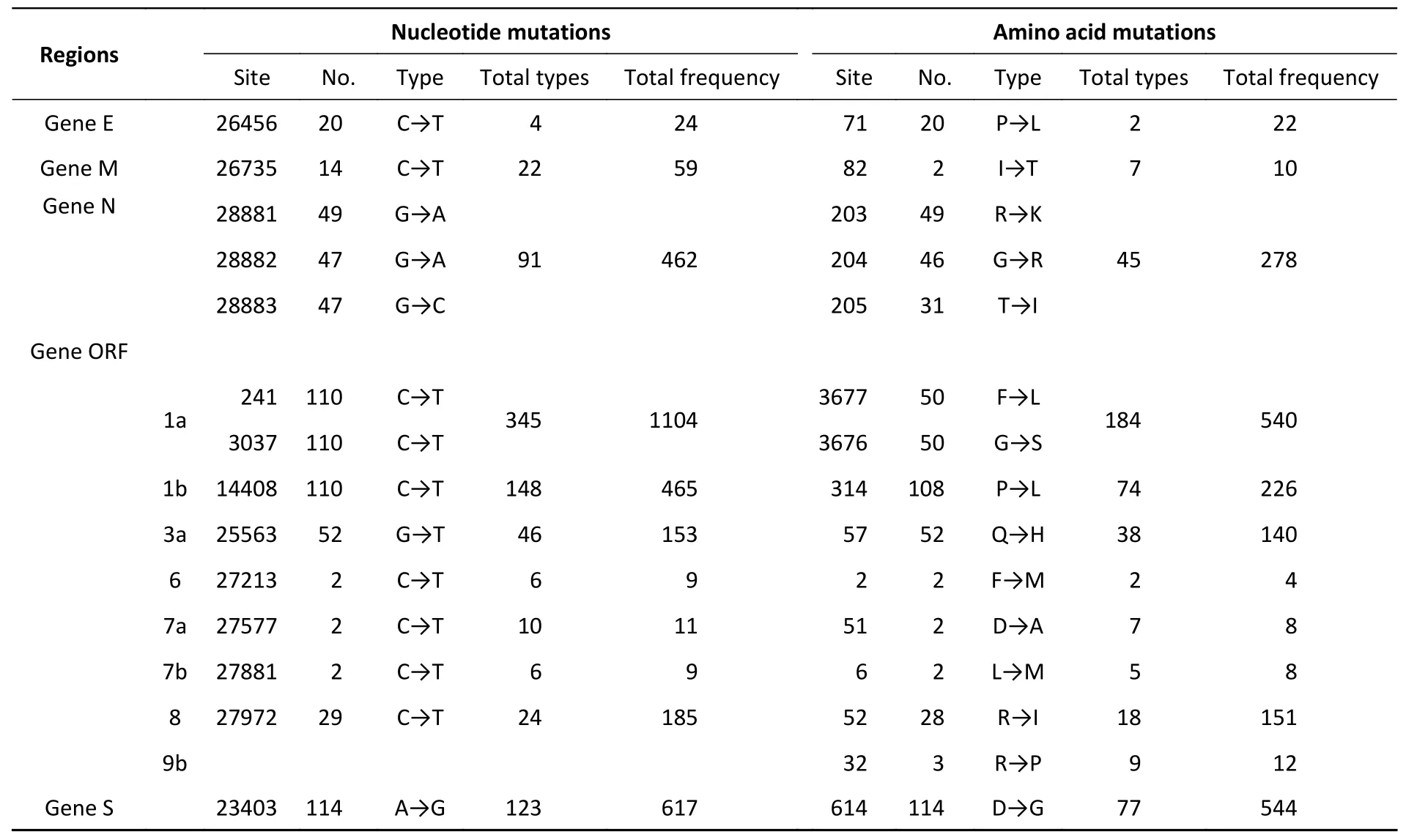
Table 2.The main location of nucleotide or amino acid variant in SARS-CoV-2 patients
DISCUSSION
We narrated the complete sequences of 117 patients with SARS-CoV-2 infection from the Guangzhou Eighth People’s Hospital,Guangdong,China,during the whole study period (June 2020 to May 2021).Based on our basic description and phylogenetic analysis of imported cases,the median age of patients in this study was younger than that of patients in earlier studies[5-7],and we noticed a close identity between the genome of the respondents and animal reference sequences[includingCanis lupusfamily,feline,mink,ferret,Panthera leo,tiger,andNeovison vison(99.76%–99.98%)].Similar findings were described previously in other studies (homology between rhinolophine,bat,pangolin,and human)[8-11].New variants with several critical mutations,such as B.1.1.7,B.1.351,B.1.617,and P.1,to which the available vaccines or potential drugs may be ineffective,emerged in 2021[12-14].The B.1.617 variant rapidly spread to India and several countriesin the world.In this study,the Indian lineage we found was sublineage B.1.618 and not the B.1.617.2 variant that was first detected and caused a pandemic in India[15].This variant was first isolated and sequenced in Guangzhou in May 2021[16].Sublineages B.1.17 and B.1.351 were considered to be the main variants.They had been affiliated with the increased transmissibility of COVID-19 epidemiology and virulence,changes in clinical disease presentation,and reduced effectiveness of available vaccines and therapeutics[17,18].In addition,we discovered the B.1.427/B.1.429,P.2,and B.1.525 variants in our studied cases. They caused community transmission or multiple clusters of COVID-19,which should arouse our attention[19-22].Previous studies showed that D614G endowed a replication advantage to SARS-CoV-2 by increasing the feasibility of human-to-human transmission[23-25].Given the limitations of epidemiological and clinical data,with a conservative interpretation method,we can only discover the D614G in the spike protein that presented the highest mutation density,presenting another limitation of our research.
According to current studies,the ORF7b gene of SARS-CoV-2 with a procaspase-3 cleavage activity has a prerequisite role in viral replication[17,26].It potentially impedes certain cellular processes that dominate several common symptoms of SARS-CoV-2 infection,such as leucine zipper formation and epithelial cell–cell adhesion,which may contribute to heart-rate disorders[26-28].The ORF9b of SARS-CoV-2,encoding a 97-amino acid protein on the mitochondrial membrane,was located within the nucleocapsid gene (N gene)[27].Previous studies revealed that SARS-CoV-2 ORF9b,as an accessory protein,inhibits innate immunity by targeting mitochondria. It can potentially suppress the antiviral response of infected cells and regulate the host immune response by suppressing type I interferon synthesis[29-31]. Among the various missense mutations in the 117 sequences,we disclosed some missense mutations in several sublineages (such as B.1,B.1.1.25,B.1.1.7,B.1.2,B.1.214.2,B.1.402,B.1.595,B.1.618,and L3) of partial lineages,including those from North America,Europe,Asia (excluding China),and Africa.They had neither been reported in the GISAID database nor published on PubMed and included five amino acid substitutions of the non-S positions and eight substitution sites in the S position.Given our lack of access to the information on clinical symptoms and dissemination chain in patients infected with the corresponding sublineages,we cannot prove whether these novel mutations contribute to its pathological functions (like serious symptoms:loss of speech or mobility,or confusion),spread,or adaptation to particular surroundings,including the age,pre-existing medical conditions,and living environment of the host.Our findings nevertheless provide important resources for tracking SARS-CoV-2 single-nucleotide variations,lineages,and clades in the imported cases of Southern China.
CONCLUSION
These imported cases in Guangdong,China,were identified,sequenced,and characterized epidemiologically and genetically,portraying the current studies on the molecular evolution of SARSCoV-2.The descriptive epidemiology approaches demonstrated the distribution of this virus.Our phylogenetic and homology analyses increased the awareness of the detailed description of this virus.Based on the results of our viral sequence analysis,sublineage diversity and high homology at the amino acid level were present in the imported cases in Guangdong.This finding confirmed the result of a previous study reporting that SARS-CoV-2 exhibited genetic diversity over time[32].All our results provide an important step in the search for viral variants with reduced pathogenicity or without potential pathogenicity.
AUTHOR CONTRIBUTIONS
No potential conflicts of interest were disclosed.LIANG Dan performed the experiment,drew figures,wrote the manuscript,analyzed phylogenetic relationships,completed statistical tables,and interpreted the results.WANG Tao experimented and undertook fragment assembly.ZHANG Guan Ting and LIANG Yu Feng were in charge of data analysis and statistics and performed the literature search and the experiment.HONG Wen Shan and LI An An performed the experiment and recorded the results.LI Jia Jiao,GUAN Da Wei,DENG Xiao Ling,CHEN Feng Juan,PAN Xing Fei,and LEI Chun Liang were responsible for collecting samples and collating the basic information about patients.WANG Li and CHEN Meng Lin gave advice on the diagrams in this manuscript.JIA Hong Ling was in charge of editing the English text of a draft of this manuscript.KE Chang Wen designed the whole study framework and took responsibility for the integrity and accuracy of the data.
CONFLICT OF INTEREST
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
The authors wish to thank Dr.LI Peng (Chinese PLA Center for Disease Control and Prevention,Beijing 510030,China) for his guidance on the diagrams in our manuscript.
Received:November 29,2021;
Accepted:March 29,2022
 Biomedical and Environmental Sciences2022年5期
Biomedical and Environmental Sciences2022年5期
- Biomedical and Environmental Sciences的其它文章
- Application of Nanopore Sequencing Technology in the Clinical Diagnosis of lnfectious Diseases*
- A Case Series of Olfactory Dysfunction in lmported COVlD-19 Patients:A 12-Month Follow-Up Study*
- Assessment of the Benefits of Targeted lnterventions for Pandemic Control in China Based on Machine Learning Method and Web Service for COVlD-19 Policy Simulation*
- lnhibition of Ciliogenesis Enhances the Cellular Sensitivity to Temozolomide and lonizing Radiation in Human Glioblastoma Cells*
- Association between Polymorphisms in Telomere-Associated Protein Genes and the Cholinesterase Activity of Omethoate-Exposed Workers*
- MiR-663a lnhibits Radiation-lnduced Epithelium-to-Mesenchymal Transition by Targeting TGF-β1*