
电石渣-矿渣复合胶凝材料性能研究
2022-06-15 14:25万宗华张文芹刘志超王发洲
硅酸盐通报 2022年5期
万宗华,张文芹,刘志超,王发洲
(1.武汉理工大学硅酸盐建筑材料国家重点实验室,武汉 430070;2.武汉理工大学材料科学与工程学院,武汉 430070)
0 引 言
电石渣是工业生产聚氯乙烯过程中电石水解产生的强碱性废渣,其主要成分是Ca(OH)2。由于电石渣的碱度较高,并且含有一些难以处理的硫化氢等有害组分,因此电石渣的综合利用率不高,若填埋或者堆存处理,又会造成土地盐碱化,严重破坏生态环境,如何实现工业废物的二次利用,是当前亟需解决的问题[1-2]。
电石渣钙质资源丰富,并且具有颗粒分散性好、比表面积大、孔隙结构大、热分解温度低等优点,在建材行业可以作为替代石灰石的二次资源,广泛用作内墙涂料、绝热材料等[3]。由于电石渣钙质资源丰富,因此可与CO2碳化反应生成CaCO3,电石渣碳化反应的机理如下:Ca(OH)2在一定的水分作用下吸收CO2气体生成CaCO3,进而形成具有较强凝聚力的结晶体(方解石为主),从而提高电石渣碳化后的力学性能[4]。但Ca(OH)2孔隙率较大,碳化反应生成的CaCO3粒径过小且易形成粒径分布较宽且不均匀的团聚体,即单一的碳化反应无法使材料获得较高性能[5]。电石渣的主要成分是Ca(OH)2,其饱和溶液pH值高达13,因此可以作为碱激发剂用于制备碱激发材料[6]。Ca(OH)2的激发效果较差,一般与其他激发剂复配使用[7],如果想单纯使用电石渣作为激发剂,胶凝材料的选用以及碱激发材料的处理就非常关键,这也是本文要重点解决的问题。
矿渣自身不具有水硬性胶凝性能,但与水和碱性激发材料充分混合反应后具有水硬性胶凝性能,这是因为在碱性环境下硅铝质矿物原料活性大,短时间内就可以产生胶凝材料,使矿渣能够脱离普通硅酸盐水泥,单独作为胶凝相材料使用[8]。一方面,Ca(OH)2作为一种常用的碱激发剂,可以通过提升矿渣前期水化放热量从而促进矿渣的水化反应,但其碱激发效率低于NaOH等常见碱激发剂[9],在实际工程应用中所需量较大;另一方面,有研究表明,大掺量的Ca(OH)2会导致抗压强度倒缩,对材料后期力学性能发展产生不利影响[10]。同时,电石渣整体粒径较小,活性较高,若能将碱激发剩余的电石渣和CO2进行合理化利用,便可实现资源的循环利用。
将过量的电石渣与矿渣混合,其中部分电石渣作为碱激发剂,另一部分电石渣与CO2反应生成CaCO3,这样既可以高效利用电石渣和矿渣等固废材料,也可以得到具有优异性能的电石渣碱激发矿渣复合胶凝碳化材料。
1 实 验
1.1 原材料
原材料包括:电石渣(CS),河南神马尼龙化工有限责任公司,电石渣的平均粒径为10.12 μm,密度为2.1 g/cm3,含水率ω=30%,将块状的电石渣放入105 ℃烘箱中烘至恒重,之后使用玛瑙研钵进行粉磨后过80目(178 μm)筛处理;矿渣,马鞍山钢铁股份有限公司,平均粒径为11.25 μm;粉煤灰(FA),福建金牛水泥有限公司,Ⅱ级粉煤灰,平均粒径为21.08 μm;偏高岭土(MK),上海昊弗化工有限公司,平均粒径为1.58 μm。电石渣、矿渣、粉煤灰和偏高岭土的主要化学成分如表1所示。
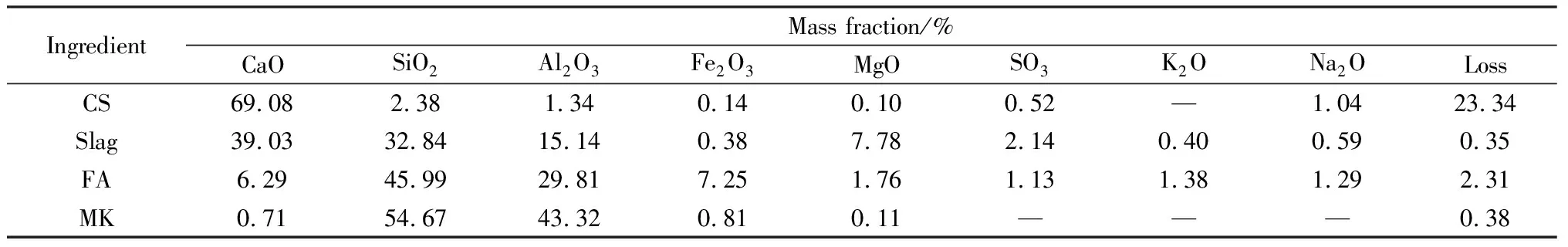
表1 电石渣、矿渣、粉煤灰和偏高岭土主要化学组成Table 1 Main chemical composition of carbide slag, slag, fly ash and metakaolin
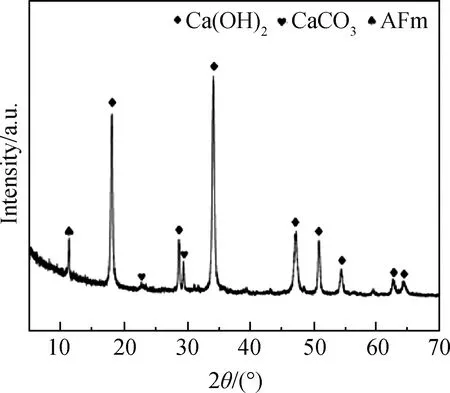
图1 电石渣的XRD谱Fig.1 XRD pattern of CS
体系中引入大量电石渣粉导致减水剂减水效果较差,工作性不好,通过引入较大粒径的粉煤灰可以很好地调控其工作性,偏高岭土中含有大量的Al/Si,可以促进碱激发反应生成C-(A)-S-H凝胶。
电石渣的XRD谱如图1所示,XRD谱中存在CaCO3、Ca(OH)2、AFm衍射峰。由于原料中含有大量水分,出现了少量碳化现象,因此XRD谱中有CaCO3衍射峰的存在。
为提升电石渣-矿渣材料的工作性,提升浇筑过程中浆体的流动性,本研究选用萘系减水剂(SP,武汉苏博新型建材有限公司),固含量为40%,减水剂中ω(Na2SO4)=6%。
1.2 试验方案
1.2.1 胶凝材料配合比对电石渣碱激发矿渣材料性能影响
设计四组电石渣碱激发矿渣胶凝体系,水灰比为0.35。胶凝体系中包括矿渣、粉煤灰、偏高岭土以及碱激发剂电石渣,搅拌时间为5 min,试块规格为20 mm×20 mm×20 mm,成型24 h后脱模,分别养护至3 d、7 d、28 d(温度为(20±2) ℃,相对湿度>95%)。试验所用配合比如表2所示。
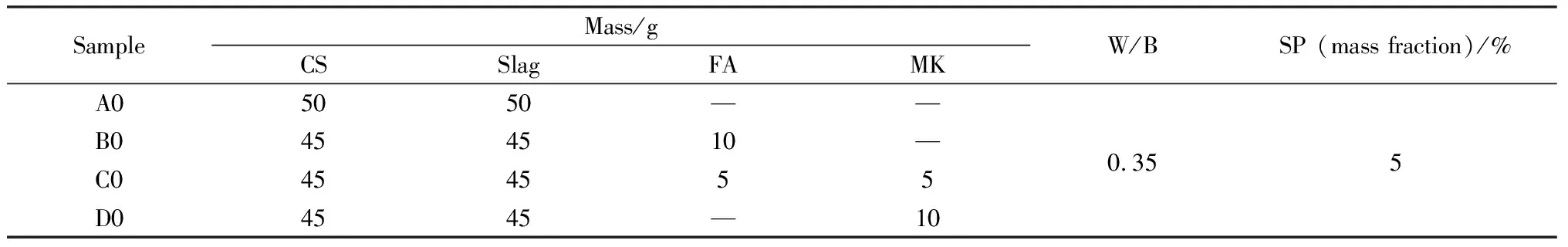
表2 电石渣碱激发矿渣净浆配合比Table 2 Mix ratio of CS alkaline activated slag paste
1.2.2 碳化制度对电石渣碱激发矿渣材料性能影响
为了保证电石渣能够在一定程度上碱激发胶凝材料,因此先碱激发至基体产生一定强度后再选择碳化(常压碳化情况下,不关闭排气口,保持压力罐内CO2气氛,排气口连接橡胶管,将CO2排入水中,通过橡胶管产生的气泡速率判断CO2流量)。在碳化试块时,将CO2气体通入压力罐中,调节压力罐分压,碳化时间为8 h。部分试块养护至3 d、7 d后取出,碳化处理完毕后继续放入标准养护室养护至28 d。
1.3 试验方法
在电石渣碱激发矿粉试块抗压强度试验中,使用三个试块抗压强度平均值作为抗压试验结果,测试时加载速率为600 N/s。
采用德国耐驰公司的STA449F3型综合热分析仪对试块进行热分析,试块在热分析前放入40 ℃真空干燥箱烘干处理。因为胶凝材料体系产物中有凝胶、CaCO3,还可能存在未反应的Ca(OH)2,因此将温度范围设定为室温至1 000 ℃,升温速率为10 ℃/min,使用N2作保护气氛。
通过计算电石渣碱激发矿渣材料TG曲线中350~500 ℃(Ca(OH)2分解)、550~750 ℃(CaCO3分解)区间的质量损失来计算试样中Ca(OH)2和CaCO3含量,计算公式如式(1)、(2)所示[11]:
(1)
(2)
式中:M1为350~500 ℃区间的质量损失;M2为550~750 ℃区间的质量损失。
采用MesoMR12-060H-1低场核磁共振仪检测试块空隙率和孔径分布规律。测试前,将试块沿棱长一半长度位置(10 mm)切割,真空保水8 h。测试时选用25 mm磁体线圈探头。回波个数(NECH)、重复采样时间间隔(TW)和累计采样次数(NS)分别为3 000、3 000和64。通过测试氢原子信号,得到周围原子环境对其弛豫时间的影响。汪鹏[12]认为,弛豫时间与孔隙尺寸之间呈正相关,所以可以在不计算孔径尺寸大小的情况下,通过比较弛豫时间的分布探究样品孔径变化规律。
2 结果与讨论
2.1 电石渣碱激发矿渣胶凝材料性能研究
根据电石渣化学成分分析,电石渣内部含有大量的Ca(OH)2(ω=91%),饱和溶液上层清液pH值为12.6,因此电石渣具有较强碱性,具备做碱激发材料的可能。何彤彤[11]认为,在碱激发矿渣胶凝体系中加入Ca(OH)2,Ca2+一部分进入孔溶液中,另一部分形成无定形C-(A)-S-H凝胶,因此Ca(OH)2具备一定的碱激发效果,但是其激发效果较弱,需与其他激发剂复配使用。为了能够充分发挥Ca(OH)2碱激发性能,本文选用过量Ca(OH)2作为碱激发剂,另选用活性较好的矿渣作为胶凝材料,其配合比见表2。
图2为电石渣碱激发矿渣胶凝体系的XRD谱,从图中可以看出,经过7 d养护后该体系内还存在大量未反应的Ca(OH)2,并且存在少量的AFm(电石渣中含有少量的S,主要以硫化物的形式存在,减水剂中也含有少量的Na2SO4,都可以与反应体系中溶出的Ca2+、Al3+反应生成AFm或AFt)。因为AFm的形貌与电石渣中的Ca(OH)2比较相近,均为片状结构,所以在通过SEM难以区分。通过对比图2(a)与图2(b)发现,随着样品龄期的增长,Ca(OH)2的衍射峰强度略微降低,这是因为电石渣中的Ca(OH)2与矿渣等胶凝材料发生反应,消耗体系内Ca(OH)2,说明过量的电石渣可以很好地激发矿渣等胶凝材料。对比3 d、7 d的XRD谱发现,7 d试块中依然存在大量未反应的Ca(OH)2,这说明体系中的Ca(OH)2过量。但通过SEM测试也发现7 d时凝胶的量明显比3 d多得多,因为凝胶为无定型相,结晶性较差,很难从XRD谱上直接显示[13]。而加入偏高岭土或者粉煤灰的试块,其Ca(OH)2衍射峰要比纯矿渣低,这说明加入偏高岭土(D0组)或粉煤灰(B0组)有利于电石渣的碱激发反应,Ca(OH)2对偏高岭土的碱激发作用比粉煤灰更明显。
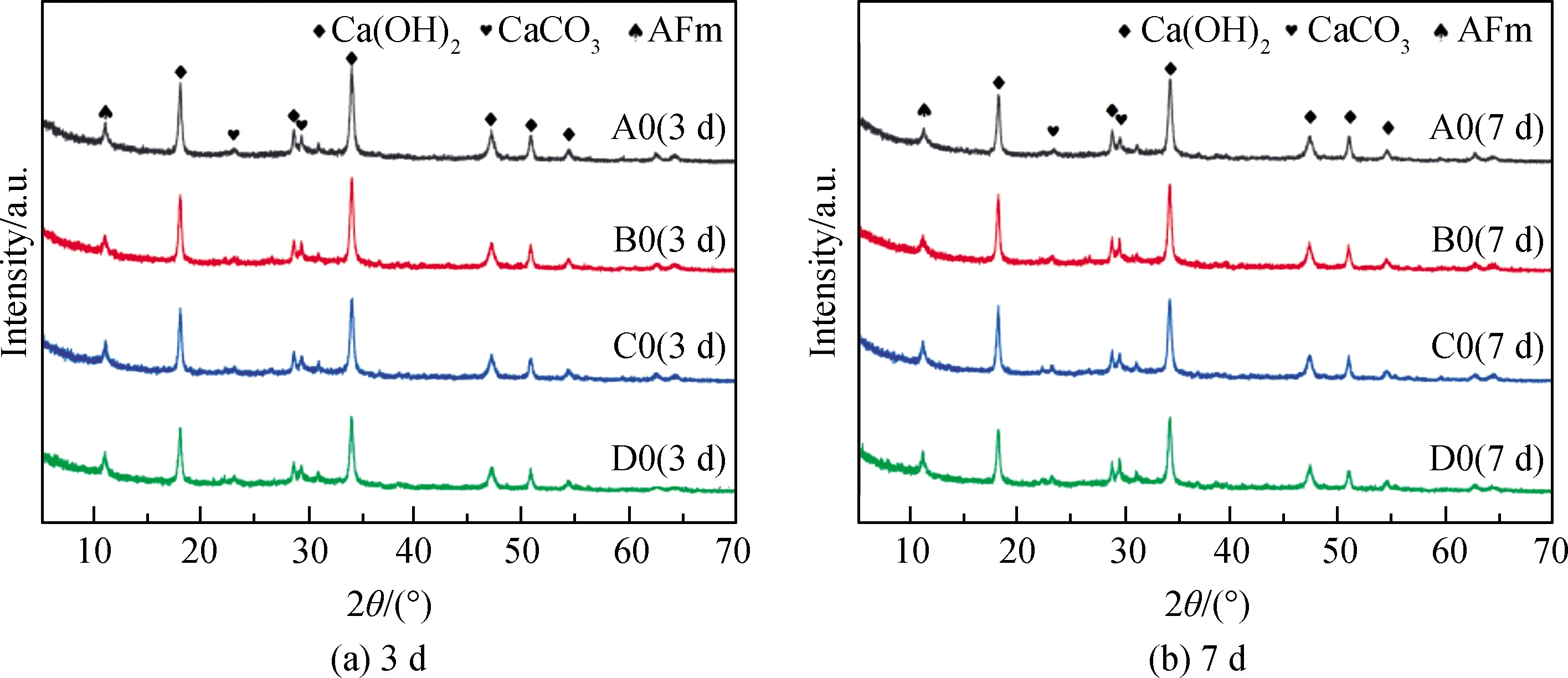
图2 电石渣碱激发矿渣胶凝体系XRD谱Fig.2 XRD patterns of CS alkaline activated slag cementitious system
为了探究电石渣对该体系的碱激发效率,加入A0组的对照组W(电石渣15%矿渣85%)进行试验。电石渣碱激发矿渣胶凝体系的水化放热曲线如图3所示。对比A0组和W组可以看出,当Ca(OH)2含量增加时,A0组的水化放热峰提前,累计水化放热提高,说明加入过量的Ca(OH)2有利于提高矿渣的碱激发效果。而加入少量粉煤灰后,粉煤灰的活性比矿渣低,导致放热峰和累计放热量都略微降低。而加入偏高岭土后,放热速率曲线出现了明显双峰,靠前的主峰是矿渣的主放热峰,而靠后的峰则是偏高岭土的放热峰,这说明电石渣对矿渣的激发效果最快,偏高岭土次之,粉煤灰最弱。累计放热量则显示偏高岭土的加入有利于提升电石渣的碱激发效果。
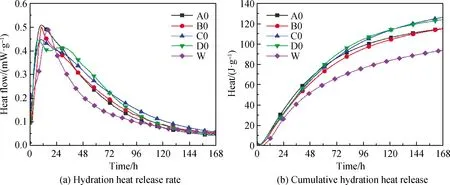
图3 电石渣碱激发矿渣胶凝体系的水化放热曲线Fig.3 Hydration exothermic curves of CS alkaline activated slag cementitious system
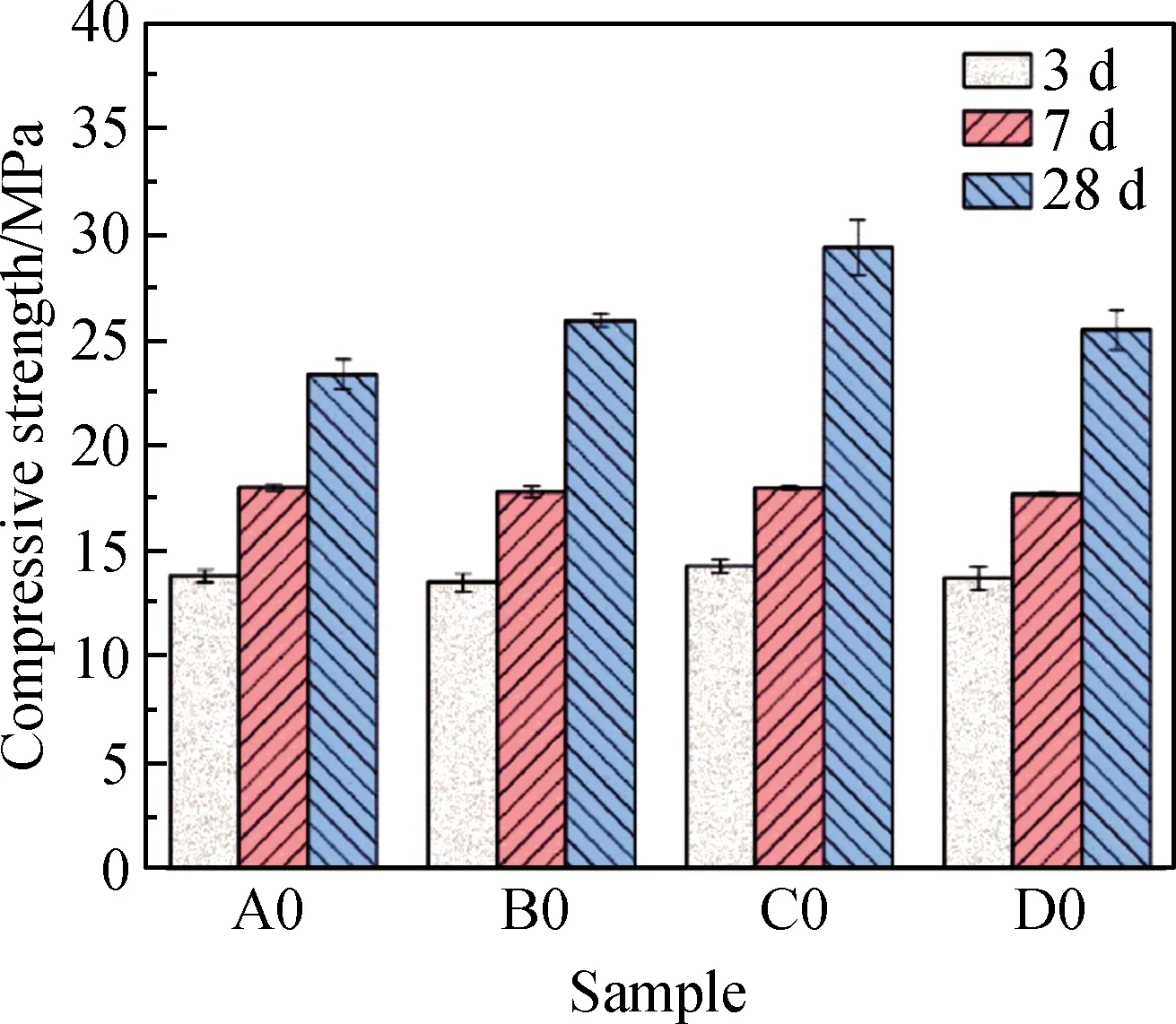
图4 电石渣碱激发矿渣胶凝体系抗压强度Fig.4 Compressive strength of CS alkaline activated slag cementitious system
图4是电石渣碱激发矿渣净浆样品在标准养护3 d、7 d及28 d时的抗压强度。随着龄期增长,试块的抗压强度明显增加,这说明Ca(OH)2对胶凝材料体系的激发较为缓慢,会持续较长时间,因此试块强度发展缓慢。而掺入粉煤灰或者偏高岭土对3 d、7 d的强度影响不大,这是因为试块结构中存在大量孔隙,结构疏松,反应生成的凝胶量虽然随着龄期有一定增加,但还无法完全密实基体,对强度无法产生明显影响。但随着龄期继续延长,碱激发反应持续进行,凝胶量继续增加,逐步密实基体,强度将持续增长。
在3 d、7 d龄期,胶凝材料的组成对体系的抗压强度影响较小,但在28 d龄期时,无论是引入粉煤灰还是偏高岭土均对强度有提升,但提升机理不同。在基体中引入粒径较大且颗粒比较圆润的粉煤灰能有效改善基体拌和时的工作性,更有利于浇筑。而且粉煤灰相对于矿渣比较惰性,即便有大量的Ca(OH)2存在,体系也只能在中后期才能发生火山灰反应,因此粉煤灰的主要作用是提高浆体工作性,使基体中各组分分布更均匀,对28 d强度仅有少量提升。而引入偏高岭土的目的是在基体中引入更多的Al/Si,从而影响产物微结构,但偏高岭土颗粒粒径较小,需水量大,无法大量引入。掺入少量粉煤灰及偏高岭土的复配C0组抗压强度最高,较空白组A0提升了25.59%,同时,具有较小粒径的偏高岭土与较大粒径的粉煤灰的加入优化了体系的颗粒级配,其中活性Al相(EDS能谱表征含Al)的溶出更是促进了C-(A)-S-H凝胶的形成,导致体系在28 d具有较好的力学性能。
图5为低场核磁谱图,主要用来表征基体孔径变化规律。由于Ca(OH)2晶体属于三方晶系,形貌为六方片状,无法紧密堆积[14],导致试块内部疏松多孔,因此基体中会出现大量的孔隙。其次因为矿渣反应生成了大量凝胶,在基体中会出现大量的凝胶孔,在图5中可以发现凝胶孔信号较高,而大孔信号较弱。对比图5中不同组分的弛豫时间谱图发现,各组基体孔径大小具有明显差异,7 d龄期试块相比3 d龄期,养护时间增加,试块内部生成更多的C-(A)-S-H凝胶,因此大孔数量减少,凝胶孔数量增加。A0组凝胶孔信号弱于C0组,A0组大孔信号强于C0组,推测这是因为体系中引入偏高岭土后含有较多的Al,碱激发时这些Al进入体系生成C-(A)-S-H凝胶,密实了基体;粉煤灰的粒径相比电石渣更大,与偏高岭土粒径级配较好。粉煤灰的反应活性较差,相比A0组,C0组在后期才发生反应密实基体。电石渣对粉煤灰和偏高岭土等掺合料都具有一定的碱激发作用,产生了C-(A)-S-H凝胶,存在大量凝胶孔,这也解释了图4中C0组的力学性能较其余各组更高。总体来说,A0组和C0组在7 d龄期时,依旧存在大量大孔,这是影响样品力学性能的主要因素。
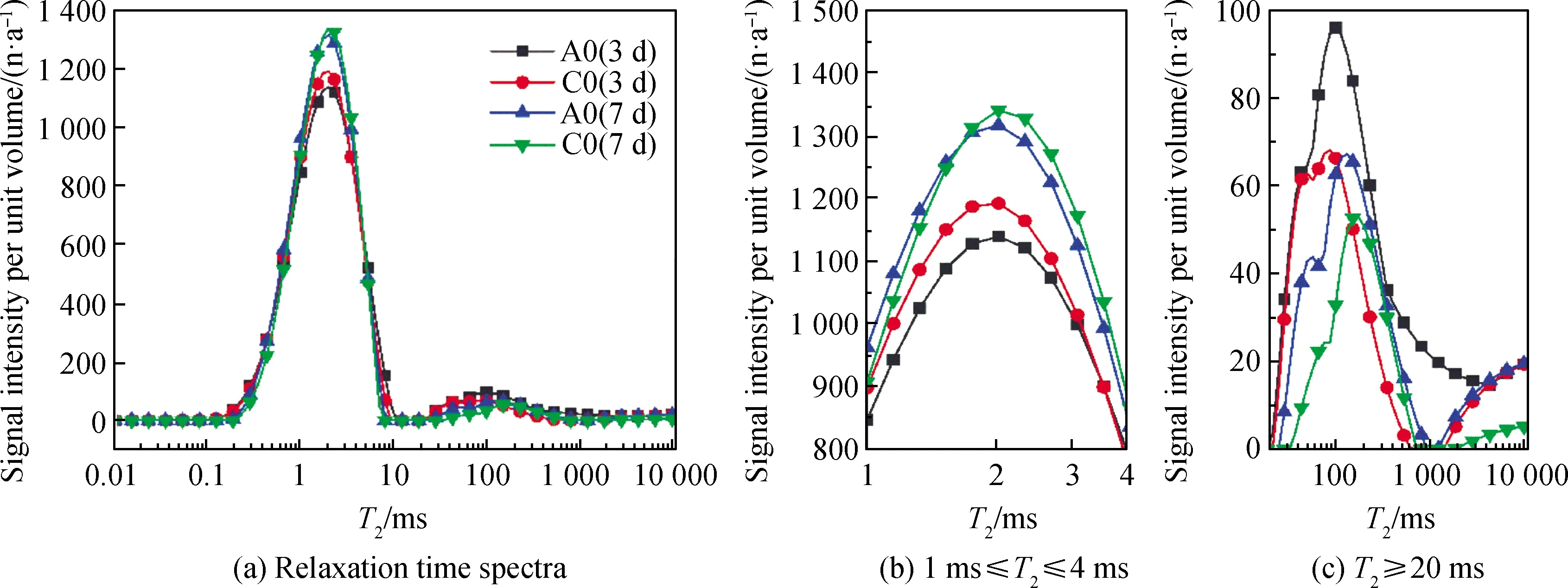
图5 电石渣-矿渣胶凝体系弛豫时间谱图Fig.5 Relaxation time spectra of CS-slag cementitious system
图6为A0组和C0组3 d、7 d龄期的SEM照片,从图中可以看出基体中存在大量未反应的不规则松散堆积的片状Ca(OH)2晶体,以及碱激发反应产生的大量C-(A)-S-H凝胶。Ca(OH)2相对矿渣具有较小的晶体粒径,导致大量的Ca(OH)2分布在矿渣间隙中(见图6(d))。在电石渣碱激发矿渣过程中,仅有部分Ca(OH)2同矿渣反应填充孔隙,导致体系中未接触矿渣的Ca(OH)2之间仍有较大空隙,使体系具有较大的孔径分布。

图6 电石渣-矿渣胶凝体系SEM照片Fig.6 SEM images of CS-slag cementitious system
2.2 碳化制度对电石渣碱激发矿渣胶凝体系的影响
对电石渣碱激发矿渣的性能进行分析,发现制约该体系强度增长的根本原因在于电石渣-矿渣基体中有大量未反应完全的Ca(OH)2。一方面,这些未反应的Ca(OH)2不规则分布造成局部存在较大缺陷,另一方面由于Ca(OH)2抗侵蚀性较差,对电石渣碱激发矿渣胶凝体系的耐久性带来较大影响。Ca(OH)2易与CO2发生反应生成稳定的CaCO3,从而消除Ca(OH)2造成的体系不稳定影响[15]。
根据以上结论,在碱激发的基础上引入碳化反应增强基体强度,选取最佳配比组C0和空白对照组A0进行碳化制度研究,两组电石渣碱激发矿渣胶凝体系所用的不同碳化制度如表3所示。
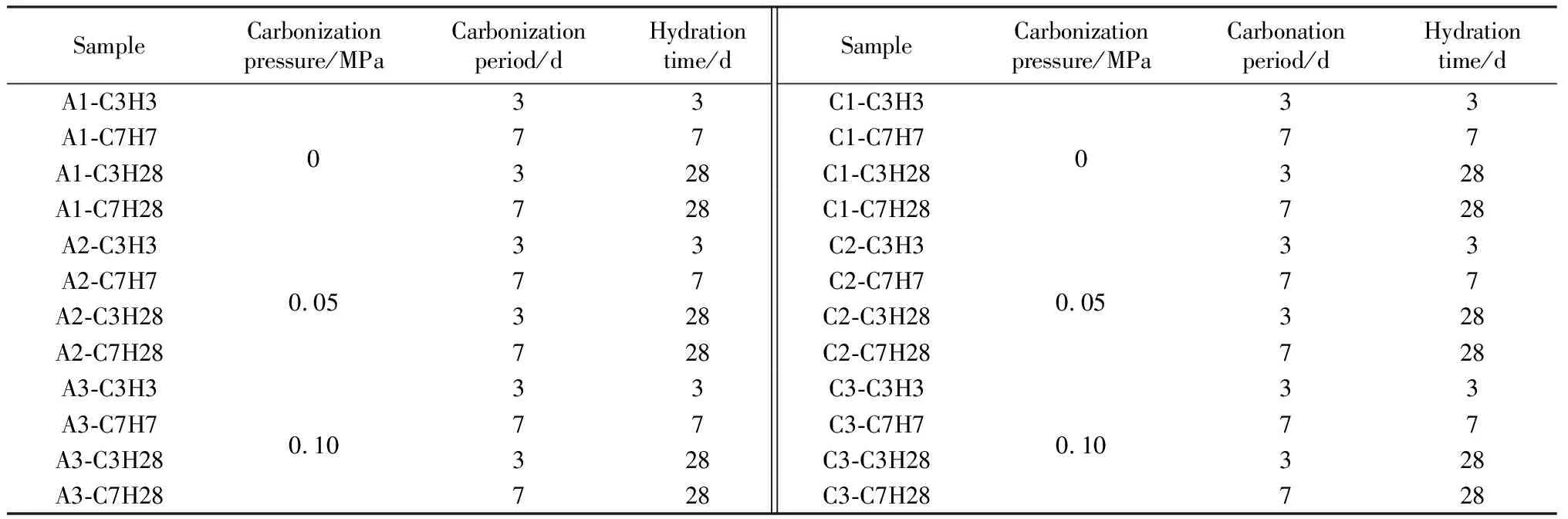
表3 电石渣碱激发矿渣胶凝体系碳化制度Table 3 Carbonization system of CS alkaline activated slag cementitious system
图7为不同碳化制度的电石渣碱激发矿渣胶凝体系XRD谱,结合图2电石渣-矿渣材料XRD谱分析发现,29.4°处CaCO3衍射峰强度明显增强,23.1°处CaCO3衍射峰强度增幅较小;Ca(OH)2的衍射峰强度降低,这是因为经过碳化处理后,电石渣-矿渣胶凝体系中部分Ca(OH)2与体系内胶凝材料发生碱激发反应,另一部分Ca(OH)2与CO2反应生成CaCO3。但是图7显示,即使经过碳化后基体中依然存在大量未反应的Ca(OH)2,碳化的程度跟基体的孔隙率密切相关,这是因为控制碳化反应的过程就是控制CO2的扩散速率。根据图5中低场核磁孔径分布,基体中存在大量孔隙,有利于CO2的扩散。为了进一步探究基体中产物分布,图8给出了试块的TG-DTG曲线图。
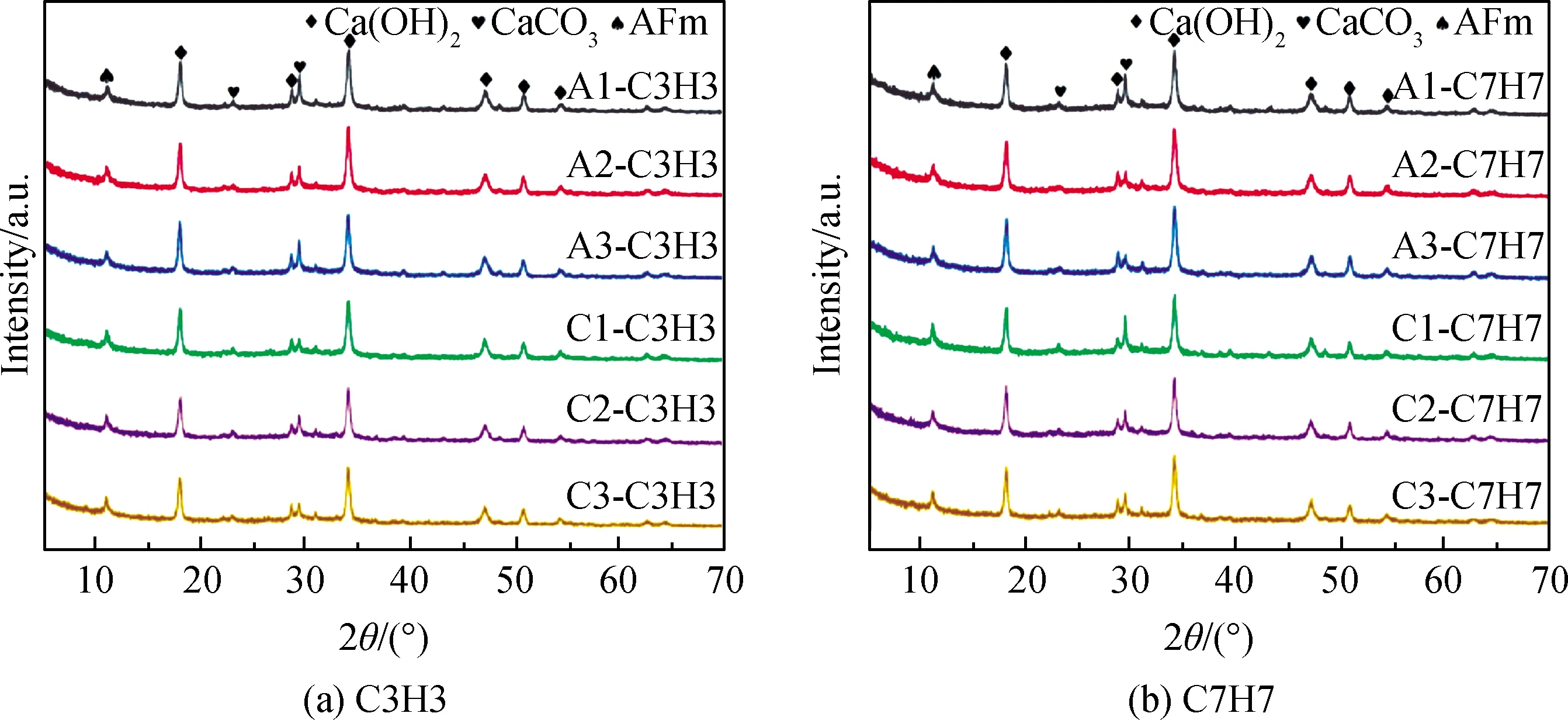
图7 不同碳化制度电石渣碱激发矿渣胶凝体系XRD谱Fig.7 XRD patterns of CS-slag cementitious system under different carbonization systems
观察图8电石渣-碱激发胶凝体系TG-DTG曲线,50~300 ℃失重对应试块内AFt(AFm)与C-(A)-S-H凝胶的分解,350~500 ℃样品失重对应Ca(OH)2分解,550~750 ℃样品失重对应CaCO3分解。Ca(OH)2与CaCO3含量由式(1)和式(2)计算得出,具体如表4所示。28 d时,根据公式(1),A组试块内剩余Ca(OH)2约19.89 g,C组试块内剩余Ca(OH)2约15.15 g。
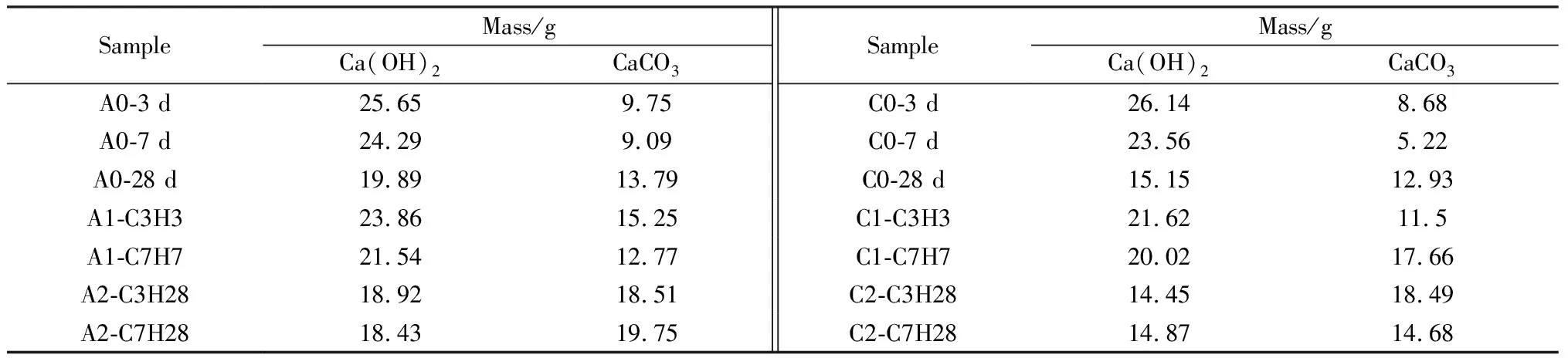
表4 电石渣-矿渣材料中Ca(OH)2及CaCO3含量Table 4 Content of Ca(OH)2 and CaCO3 in CS-slag material
试块在压力罐中的碳化过程为:CO2气体逐渐向试块内部扩散,试块外部的Ca(OH)2迅速碳化生成致密的CaCO3层,使得CO2气体后期不易进入试块内部,导致试块内部碳化不均匀,Ca(OH)2未完全反应。试块承压时,主要由试块外围承受压力,试块实际受压面减小,因此抗压强度降低,所以在相同碳化时间下,低分压条件下碳化的试块抗压强度更高。
图9为不同碳化制度下电石渣碱激发矿渣净浆的抗压强度,虚线为对应配比相应龄期碳化前的强度。由图9可知,3 d和7 d时,碳化对电石渣-矿渣胶凝体系力学性能有提升作用,其中A1组和C1组经常压碳化处理后抗压强度最高,说明碳化分压最低,电石渣-矿渣胶凝体系力学性能提升越明显,这与乔欣元[16]所得结论一致。
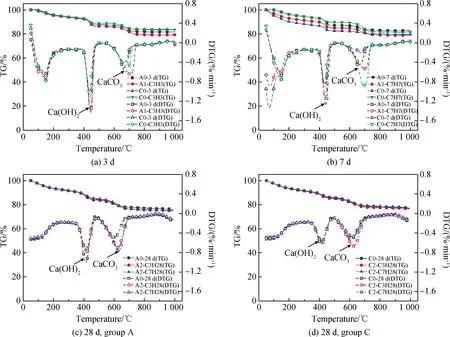
图8 电石渣-矿渣胶凝体系热分析曲线Fig.8 Thermal analysis curves of CS-slag cementitious system
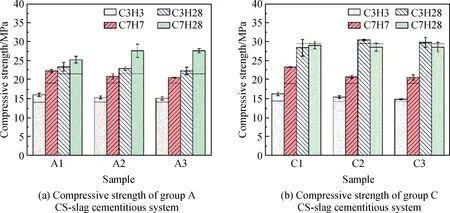
图9 不同碳化制度下电石渣-矿渣胶凝体系的抗压强度Fig.9 Compressive strength of CS-slag cementitious system under different carbonization systems
对比不同碳化制度试块发现,A组在7 d后碳化并养护至28 d时力学性能最佳,当碳化分压为0.05 MPa与0.10 MPa时,试块抗压强度最高且相近,相比于无碳化处理试块(A0组)有明显提升,提升了23.56%。C组经碳化并养护至28 d时力学性能相近,其中C组在3 d时经碳化分压0.05 MPa处理后,28 d抗压强度可达30.49 MPa。
碳化对电石渣-矿渣试块(A组)3 d、7 d和28 d强度均有一定提升,这是因为A组内部大孔较多,有利于CO2向试块内部扩散,使强度增长。图10为碳化电石渣-矿渣胶凝体系SEM照片,SEM测试分析显示CO2的扩散深度是十分有限的,仅能在表面接近10 μm处(见图10(a)、(b))观测到明显的碳化产物层,从图10(c)可以看出,试块内部仍然以C-(A)-S-H凝胶为主,以及少量的碳酸钙与一些未反应完全的Ca(OH)2。而C组只在3 d和7 d时,力学性能有一定提升,28 d时强度无明显增长。推测是由于28 d时,C组体系的碱激发效果更好,基体本身相对于A组更加致密,大孔数量减少,表面形成了更加致密的碳酸钙硅凝胶层,碳化处理未深入试块内部,只停留在试块表面,所以养护到28 d时,与碳化作用相比,电石渣碱激发作用对于基体的力学性能提升更明显。
对比基体内部中心部位与基体表面形貌发现,试块表层生成了高致密性CaCO3产物层,虽然CaCO3产物层的强度较高,但阻碍了CO2气体扩散进入基体内部,进而阻碍了剩余Ca(OH)2发生碳化反应,影响了试块抗压强度的提升。对比图10(c)~(f)发现,与3 d时相比,养护龄期为7 d时,试块内部凝胶更多,电石渣碱激发矿渣反应更充分,凝胶包裹CaCO3后连结了纳米粒径大小的CaCO3填充在试块内部空隙,提升了试块强度。

图10 碳化电石渣-矿渣胶凝体系SEM照片Fig.10 SEM images of CS-slag cementitious system
根据表4中Ca(OH)2及CaCO3含量可以看出,试块经过不同碳化制度处理养护至28 d时,内部依然有较多的Ca(OH)2未反应,因此28 d强度增长一方面来源于碱激发,另外一部分强度来源于表层的致密碳化层。若想提高试块的强度必须解决内部未反应的Ca(OH)2,因此我们在C0组基体中引入2%(质量分数)尿素,仅使用内部碳化源的方式使内部Ca(OH)2充分碳化,密实基体。密实后基体的3 d强度为17 MPa,7 d强度可达到25 MPa。
尿素在碱性条件下分解产生CO2,与体系内未反应的Ca(OH)2发生碳化反应,生成CaCO3填充在试块内部空隙,密实基体。因此掺入尿素后基体的3 d和7 d强度相对于C0组有明显提升。其中7 d提升幅度较大,这可能是因为7 d时碳化反应产生的碳酸钙与胶凝材料体系中的凝胶结构产生了较好的交联结构,从而提升了基体力学性能,观察掺入尿素的SEM照片(见图11)可以验证上述观点。

图11 内部碳化电石渣-矿渣胶凝体系SEM照片Fig.11 SEM images of internal carbonization CS-slag cementitious system
无论是外部碳化还是内部碳化都可以提升基体强度,但加入少量的尿素无法完全消体系内部Ca(OH)2,如何选取合适的内部碳化源以及如何实现内部碳化法与外部碳化法协同提升电石渣-矿渣胶凝体系的力学性能,还有待进一步探究。
3 结 论
(1)电石渣对矿渣等胶凝材料有良好的碱激发效果,其中掺入粉煤灰以及偏高岭土的电石渣-矿渣复合胶凝体系性能最优,但体系内仍留有大量Ca(OH)2,孔隙率较高。
(2)采用外部碳源法可以有效提升电石渣-矿渣胶凝体系的力学性能,但是会在表面形成致密的碳酸钙硅凝胶层,阻碍CO2进入体系内部与Ca(OH)2反应,使得体系内产物分布不均匀。
(3)在本文的原材料与试验条件下,采用内部碳源法,在体系中加入尿素同样可以提升电石渣-矿渣胶凝体系的力学性能。
猜你喜欢
河北科技师范学院学报(2022年2期)2022-08-26
中国公路(2022年10期)2022-08-03
水泥技术(2022年2期)2022-03-28
建材发展导向(2022年4期)2022-03-16
西南科技大学学报(2021年1期)2021-12-17
能源工程(2021年3期)2021-08-05
铁道建筑技术(2021年4期)2021-07-21
矿产勘查(2020年11期)2020-12-25
中国金属通报(2019年10期)2019-11-27
建材发展导向(2019年10期)2019-08-24
