
一种快速简便的伪狂犬病病毒EP0基因RT-qPCR检测方法的构建
2022-09-23 06:39张丽荣阮可悦李国新单同领童光志
中国动物传染病学报 2022年4期
张丽荣,阮可悦,童 武,李国新,高 飞,单同领,于 海,童光志,郑 浩
(中国农业科学院上海兽医研究所,上海 200241)
伪狂犬病(Pseudorabies, PR),又称Aujeszky's病,是由伪狂犬病病毒(Pseudorabies virus, PRV)引起猪、牛、羊等多种家畜和野生动物以发热、奇痒(猪除外)以及脑脊髓炎为主要症状的一种急性传染病[1]。猪是该病的主要传染源以及自然贮存宿主[2]。猪感染伪狂犬病后常引起初生仔猪发病,死亡率100%[3];育肥猪生长缓慢甚至成为僵猪;种猪丧失种用价值;妊娠母猪流产、产死胎或木乃伊胎[4-5]。与其他疱疹病毒一样,伪狂病病毒具有长期潜伏感染、终生带毒的特征。
PRV的增殖是遵循严格的时序方式进行的。Ben-Porat等根据PRV DNA转录和表达时间的先后顺序将基因分为立即早期基因(immediate early gene,IE),早期基因(early gene, E)和晚期基因(late gene, L),并以级联方式进行调控[6]。PRV只有一个立即早期基因,即IE180[7]。早期基因有EP0基因、TK基因以及UL54基因。其中EP0基因位于UL区,其转录方向与潜伏相关转录体(large latency transcripts,LLT)方向相反,与立即早期基因IE180彼此相邻且转录方向一致。由于EP0基因与反转录的LLT大部分重叠,缺失LLT基因的同时难以避免缺失一部分的EP0序列,从而影响EP0的表达[8]。
EP0转录的mRNA约为1.75 kb,分子量为45 kDa左右,可编码1230个核苷酸(编码410个氨基酸)的开放阅读框[9]。EP0基因是PRV的早期基因,而与之相对应HSV-1的ICP0早期基因则为极早期基因,但EP0具有ICP0蛋白相应功能。ICP0蛋白分子是HSV-1病毒极早期基因、早期基因以及晚期基因的反式激活因子,其锌指结构为反式激活动能所必需。有研究表明:EP0基因对于PRV的复制是非必需的,但是EP0作为病毒基因启动子的反式激活物,对于潜伏感染的激活是必要的[10-11]。在体外培养的细胞中,EP0可以激活基于TATA框启动子的转录起始[12]。在体内,EP0可以激活多种PRV编码基因的启动子,如IE180、UL23(胸苷激酶)和US4(gG)等[13]。
实时荧光定量PCR技术已经广泛应用于很多病毒的检测,不仅可以对检测的序列进行定性分析,而且可以对其进行准确定量,与常规PCR技术相比具有更高的敏感性、特异性和更好的重复性,以及快速和污染几率低等优点[14]。本实验室采用SYBR GreenI建立了一种PRV EP0基因的实时荧光定量检测方法,以期为EP0基因的定量分析提供一种简便快速、经济和灵敏的检测方法,同时也为PRV潜伏感染时病毒表达的研究奠定基础。
1 材料与方法
1.1 病毒 伪狂犬病病毒变异株JS-2012株为实验室分离鉴定并保存[15]。LLT单个启动子缺失病毒JSL1R1[16]、双启动子缺失病毒JS-L1R3和JS-UL1R3均由本实验构建和保存。
1.2 主要试剂 反转录试剂盒、PBS磷酸盐缓冲液、TRIzol试剂均购自中国赛默飞世尔公司;荧光定量PCR所用试剂购自TaKaRa公司;细胞培养基DMEM购自上海西格玛奥德里奇公司;培养细胞所用FBS胎牛血清、消化胰酶0.25%Trypsin-EDTA(1×)购自Invitrogen(美国)。
1.3 接毒以及RNA的提取 将接种用细胞均匀的铺在6孔板中,待细胞长至90%以上时接毒。按照1 MOI的剂量接种细胞,放置细胞培养箱(37℃、5%CO2)中吸附1 h后弃掉病毒液,换成含2%的FBS培养基继续放置细胞培养箱中培养。在接毒8 h后按照TRIzol总RNA提取液说明书提取样品的总RNA。
1.4 DNaseI消化 按照DNaseI消化试剂盒使用说明书进行操作,消化体系为25 μL:总DNA模板为21.5 μL,DNaseI消化酶1 μL,10× DNaseIReaction buffer 2.5 μL,轻轻混匀后放置37℃水浴30 min,加入0.25 μL的0.5 mol/L的EDTA,75℃水浴10 min以终止反应。
1.5 反转录过程 按照反转录试剂盒说明书进行操作,反转录总体系为20 μL:RNA模板8 μL,Random primer(0.1 μg/μL)1 μL,RNase-free water 3 μL,轻轻混匀后放置65℃水浴5 min,立即放冰上备用。然后加入5× Reaction buffer 4 μL,10 mmol/L dNTP Mix 2 μL,Ribolock RNase Inhibitor(20 U/μL)1 μL,Revert Aid M-MuL VRT(200 U/μL)1 μL,轻轻混匀后先放置25℃条件下反应5 min,之后45℃条件下反应60min,最后放置70℃条件下终止反应5 min。
1.6 RT-qPCR方法的构建以及优化 根据本实验室获得的两种不同剪接方式的EP0基因序列对比结果分析,采用Primer Premier 5.0软件设计荧光定量检测引物,EP0334-F:5-GGAAGAGGATGAG CCGGTCT-3;EP0442-R:5-GGTTCATCCCGTG CTCCTG-3。荧光定量反应体系为:2× SYBR Green PremixEx TaqTM10 μL,上、下游引物各0.4 μL,cDNA为2 μL,最后用RNase-Free water 补足至20 μL。设计3个退火温度(分别为55℃、60℃和65℃),选择反应信号强度最高,Ct值最小的退火温度作为最适反应条件。
1.7 病毒基因组对RT-qPCR方法的影响 提取病毒的RNA后,将所得样品分为两份处理,一份利用DNaseI消化处理,一份不使用DNaseI消化,检测DNaseI消化前后的RNA中EP0基因是否表达。
1.8 不同反转录引物对RT-qPCR方法的影响 分别选择反转录试剂盒中的Random Primer和Oligo(dT)对所提取的RNA进行反转录,检测不同反转录引物对EP0基因表达量的影响。
1.9 EP0基因的时态表达特征 根据JS-2012的病毒滴度按照1 MOI的剂量接种细胞,分别在2、4、8、12 h和16 h收取病毒RNA,检测EP0基因的动态表达特征。
1.10 RT-qPCR方法的初步临床应用 将JS-2012野毒株和LLT启动子缺失突变病毒按照104TCID50的剂量滴鼻接种小鼠,观察小鼠发病情况和死亡情况,并利用构建好的RT-qPCR方法检测小鼠三叉神经组织中EP0的表达量。
2 结果
2.1 荧光定量RT-qPCR构建及优化 试验结果显示:60℃退火温度条件下可获得良好的扩增效果。在这个反应条件下反应信号强度最高,Ct值最小(图1),并且溶解曲线单一、重复性较好(图2)。所以本RT-qPCR方法的最适反应条件为:95℃预变性3 min,95℃变性15 s,60℃退火30 s,共40个循环。反应体系为:2×SYBR Green PremixEx TaqTM10 μL,上、下游引物(10 μmol/L)各0.4 μL,cDNA 2 μL,用RNase-Free water补足至20 μL。
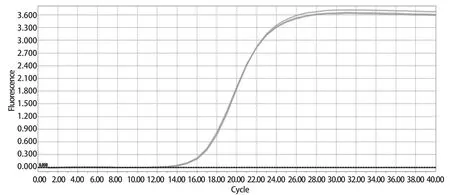
图1 RT-qPCR扩增曲线Fig.1 Amplification curve of RT-qPCR
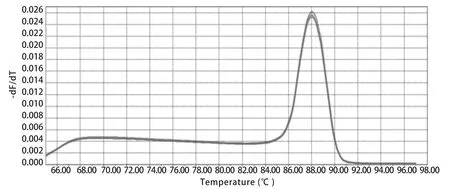
图2 RT-qPCR溶解曲线Fig.2 Melting curves of RT-qPCR
2.2 病毒基因组对RT-qPCR方法的影响 为了避免病毒基因组对反转录的影响,本研究利用DNaseI将提取的RNA进行消化。结果显示:将DNaseI处理前后的RNA分别作为模板进行qPCR,结果显示DNaseI消化前后,均检测不到EP0的转录。PRV基因组DNA为模板,也检测不到EP0的转录。但是DNaseI消化前后的RNA作为模板进行反转录得到的cDNA作为模板进行qPCR,结果显示在DNaseI消化前后均能检测到EP0的表达,并且差异不显著。这表明,病毒基因组DNA的存在与否并不影响该RT-qPCR方法的检测结果(图3)。

图3 病毒基因组对RT-qPCR的影响Fig.3 The effect of the viral genome on RT-qPCR
2.3 不同反转录引物对RT-qPCR方法的影响 为了提高反转录效率,本研究利用反转录试剂盒中提供的两种引物Random primer和Oligo(dT)分别对总RNA进行反转录,比较两种引物的反转录效率,RT-qPCR结果显示:利用Random Primer反转录,EP0的表达量显著高于Oligo(dT)反转录表达量(P<0.0001),差异极显著,具有统计学意义(图4)。

图4 不同引物对比结果Fig.4 Comparison results of different primers
2.4EP0基因的时态表达特征 将亲本病毒按照1 MOI的剂量接种6孔板,利用TRIzol试剂收取不同时间点的细胞总RNA,利用本研究建立的RT-qPCR方法检测细胞中的EP0的动态表达情况。结果显示:病毒感染后2 h,EP0开始表达,这表明EP0是一种早期基因;在8 h后EP0基因的表达量达到最高,之后随着时间的延长逐渐减少(图5)。

图5 EP0基因的时态表达特征Fig.5 Temporal expression characteristics of EP0 gene
2.5 RT-qPCR的临床初步应用 按照104TCID50的剂量滴鼻接种小鼠,结果发现突变病毒也能造成小鼠发病并死亡。在攻毒后的第3 d小鼠开始发病,出现典型的伪狂犬病临床神经症状:部分小鼠表现为异常兴奋,头部瘙痒,频繁抓挠头部皮肤导致头部抓破;部分小鼠表现为精神沉郁、畏寒,肢体蜷缩在一起,食欲减退,但最终所有小鼠均发病死亡。然后在小鼠死亡后立即取三叉神经并提取RNA反转录,利用建立好的荧光定量PCR方法对不同病毒感染的小鼠三叉神经组织进行检测。结果显示:该方法可以检测到三叉神经组织中EP0基因的表达。LLT启动子缺失突变病毒也可以表达EP0,但是与亲本病毒JS-2012相比,突变病毒中EP0的表达量大大降低。比较单个启动子缺失病毒JS-L1R1与双启动子缺失病毒JS-L1R3的表达量差异极显著(P<0.0001),这表明第二个启动子缺失后可以影响EP0的表达。比较JS-L1R3和JS-UL1R3,结果显示JS-UL1R3的表达量较少(P<0.05),这表明启动子前重复序列的缺失也会影响EP0的表达(图6)。

图6 小鼠三叉神经中EP0基因的表达水平Fig.6 Expression level of EP0 gene in trigeminal nerve of mice
3 结论
利用分子手段检测生物标志物已经成为了一种趋势,并且随着PCR技术的发展,更加精密、灵敏、高通量的PCR检测方法被开发并应用于临床监测。如数字PCR、seninest-PCR、多重荧光定量PCR等。对于含量极低、容易溶解、在蛋白水平检测时受基质影响较大,以及其他不能在蛋白水平检测的生物标志物,均可以尝试在核酸水平对其检测。
近来,我们检测到的EP0基因ORF中存在一内含子(231~368 nt),本研究根据EP0基因剪接后的序列成功建立了伪狂犬病病毒EP0基因的SYBR GreenI实时荧光定量PCR检测方法。由于提出的PRV感染细胞总RNA中存在病毒基因组DNA,分析病毒基因转录水平时,常需要DNaseI消化去除PRV DNA的残留,以获得准确的实验结果。本文建立的EP0基因定量检测方法,对PRV基因组DNA、PRV感染细胞总RNA及其DNaseI消化处理的RNA,检测结果均为阴性;但是利用引物反转录后,DNaseI消化前后的样品均能检测到EP0基因的表达,并且表达量差异不显著。这表明,PRV基因组的存在不干扰该方法的检测结果,在EP0转录时不需要DNaseI处理总RNA样品,简化了EP0转录检测的实验流程,提高了检测效率,也可节省部分成本。同时,通过比较利用随机引物和Oligo(dT)这两种引物的反转录,结果显示随机引物具有转录检测效果。我们也利用该方法检测了EP0基因在细胞中的时态表达特征,结果显示EP0基因在感染后2 h已存在转录,8 h时表达量最高,之后逐渐减少。这显示出EP0是一种早期基因。随着感染时间延长,EP0转录量出现下降,这可能与EP0具有自调控功能有关。郭洪[17]研究显示,EP0在转染的细胞中能抑制其自身启动子的活性,表现出对自身表达的负调控功能。将该方法应用于感染不同病毒株的小鼠实验中,结果显示在小鼠三叉神经组织中均能检测到EP0基因的表达,并且LAT启动子缺失后EP0的表达比亲本毒株出现显著下降。因此本研究构建的RT-qPCR检测方法可以很好的检测EP0基因的表达,并且方法简单快速,也为EP0基因的功能研究奠定基础。
猜你喜欢
军事文摘(2022年16期)2022-08-24
祝您健康(2022年2期)2022-01-14
今日农业(2021年11期)2021-08-13
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
中国报道(2018年2期)2018-04-20
小朋友·快乐手工(2017年3期)2017-04-26
饮食科学(2016年7期)2016-07-27
中学生数理化·七年级数学人教版(2014年1期)2014-06-20
