
华西牛胴体性状GWAS 分析
2022-11-17 03:52王泽昭李宏伟安炳星李海鹏徐凌洋蔡文涛张路培高会江李俊雅
中国畜牧杂志 2022年11期
高 翰,葛 菲,王泽昭,李宏伟,安炳星,李海鹏,徐凌洋,朱 波,蔡文涛,张路培,高 雪,高会江,陈 燕,李俊雅
(中国农业科学院北京畜牧兽医研究所,北京 100193)
我国是肉牛生产大国,也是牛肉消费大国[1]。随着居民生活水平的提高,消费者对牛肉的需求持续增长,供需缺口呈扩大趋势[2]。胴体性状作为肉牛生产实践中最重要的经济性状之一,是典型的由多基因调控的数量性状,遗传调控机制复杂[3-4]。Risch 于1996 年首次提出全基因组关联分析(Genome-Wide Association Study,GWAS)的应用[5],随着测序技术的不断发展和测序成本的降低,GWAS 被广泛应用于研究家畜数量性状的研究中[6-10]。利用GWAS,在肉牛中鉴定出了PLAG1[11]、IGF2[12]、MTPN[13]等影响胴体性状的重要基因。大部分GWAS 都是基于单一性状与SNPs 的关联分析,即使同时分析多个相关表型,也只是逐个性状分别进行单一性状GWAS[14-15]。Bolormaa 等[16]研究指出,对于具有高估计育种值(Estimated Breeding Value,EBV)准确性的性状,多性状GWAS 的效力至少会与单性状GWAS 的效力一样高。如果性状间高度相关,则多性状分析相较于单性状分析更具有优势。因为多性状GWAS 只进行一次统计检验,同时考虑多个性状的性状内和性状间方差组分[17],降低了多重检验造成的误差[18],提高了检验功效[19-20]和参数估计的精度[21]。因此,与逐个性状与遗传位点间的关联分析相比,多个性状联合信息的多性状GWAS 通常具有更好的关联分析效果[22-24]。
“华西牛”是中国农业科学院北京畜牧兽医研究所历经40 余年潜心培育的肉牛新品种,2021 年通过国家畜禽遗传资源委员会审定,并获得国家畜禽新品种证书,其具有生长速度快、饲料转化率高、净肉率高、产肉和繁殖性能好、抗逆性强等特性。与国际同类型肉牛品种相比,“华西牛”的日增重、屠宰率、净肉率处于国际先进水平。本研究以内蒙古乌拉盖管理区建立的华西牛资源群体为研究对象,基于Illumina BovineHD 芯片基因型数据,对胴体重(Carcass Weight,CW)、胴体长(Carcass Length,CL)、胴体胸深(Chest Depth of Carcass,CD)3 个胴体性状分别进行单性状与多性状GWAS 分析,旨在挖掘影响华西牛胴体性状的显著SNPs 位点及候选基因,为全基因组选择育种提供有效的分子标记,为进一步研究华西牛胴体相关性状的潜在遗传机制提供有价值的参考。
1 材料与方法
1.1 实验数据收集 实验动物群体选自中国农业科学院北京畜牧兽医研究所牛遗传育种创新团队在内蒙古锡林郭勒盟乌拉盖管理区组建的华西牛资源群体,该群体2008 年开始建立,每年进行扩群,并且使用Illumina BovineHD 芯片对每个个体进行基因分型。胴体重、胴体长和胴体胸深3 个胴体性状表型数据严格按照GB/T 27643-2011《牛胴体及鲜肉分割》[25]的要求进行测定。
1.2 表型处理和基因型质控 对表型数据进行处理,包括剔除表型缺失值、三倍标准差以外的异常值。采用一般线性模型评估育肥场、屠宰场、屠宰年份、屠宰季节以及年龄对性状影响的显著性,将显著的效应作为协变量加入到模型当中。利用PLINK 软件[26]对群体进行主成分分析(Principal Components Analysis,PCA),使用R 包ggplot2 绘制群体PCA 图。利用EIGENSTRAT计算各主成分是否具有显著统计学意义,将达到显著水平P<0.05 的主成分作为协变量加入到模型当中。使用PLINK 软件对相应个体的基因型数据进行质量控制,标准如下:剔除SNP 缺失率大于10%的个体以及检出率低于90%、最小哈德温伯格平衡小于1.0×10-6、最小等位基因频率小于0.05 的SNPs 位点。最后,使用R包ASReml[27]中的多性状动物模型计算各性状之间的表型相关和遗传相关。
1.3 关联统计分析
1.3.1 单性状GWAS 使用GEMMA 软件[21]进行全基因组关联分析,采用考虑亲缘关系的混合线性模型(Linear Mixed Model,LMM),计算公式如下:

式(1)中,y为所有个体表型性状的n×1 向量;W为协变量矩阵,α为包括截距在内对应系数的一列向量;x为标记基因型向量,β表示标记位点效应的大小;u为个体的随机效应向量,u~N(0,KVg),K代表由SNP 标记计算出的已知的n×n遗传关系矩阵,Vg为加性遗传方差;ε代表n×1 随机误差向量,符合ε~N(0,IVe)分布,I代表n×n身份矩阵,Ve代表残差组分。本次研究中主要利用Wald 测验作为GWAS 分析显著统计指标。
1.3.2 多性状GWAS 使用GEMMA 软件[21]中的多性状混合线性模型(Multivariate Linear Mixed Model,mvLMM)进行多个性状表型值和SNP 标记的GWAS,计算公式如下:

式(2)中,Y为n×t维表型矩阵,n是样本数目,t是分析性状个数;W为n×c维协变量矩阵,A为c维相应系数行向量,c是包含截距项在内的行变量个数;x为n维当前检验SNP 的基因型指示变量列向量,β为n×t维不包括当前检验SNP 的剩余多基因效应矩阵;G~MN(0,Vg⊗K),K是由全基因组SNP 标记构建的基因组亲缘关系矩阵,Vg是t×t维剩余多基因方差-协方差矩阵;E为n×t维误差项矩阵,E~MN(0,Ve⊗I),Ve是t×t维误差方差-协方差矩阵,I是单位矩阵。最终,全基因组上每个SNP 都能得到1 个一因多效Wald 统计量。应用Bonferroni 方法进行多重校正,确定染色体水平显著性阈值为1/N,N 为用于分析的SNPs 数目。最后,用R 包ggplot2 绘制曼哈顿图和QQ 图。
1.4 位点版本转化及基因注释 Illumina BovineHD 芯片中的SNPs 位置信息为Bos taurus UMD 3.1 版本,使用在线网站UCSC 中的liftover 工具(http://genome.ucsc.edu/cgi-bin/hgLiftOver/)将其转化成最新的牛基因组版本ARS-UCD 1.2。根据得到的显著SNPs 位点的物理位置,利用Ensemble 在线数据库的biomart 模块(http://www.ensembl.org/biomart/)搜索每个显著SNP位点上下游50 kb 区域寻找距离最近的候选基因,作为与目标性状关联的重要功能基因。
2 结果
2.1 主成分分析 由图1 可知,实验群体存在分层现象。利用EIGENSTRAT 计算发现,有2 个达到P<0.05 显著水平的主成分,因此在后续分析中将前2 个主成分作为协变量加入到模型中。
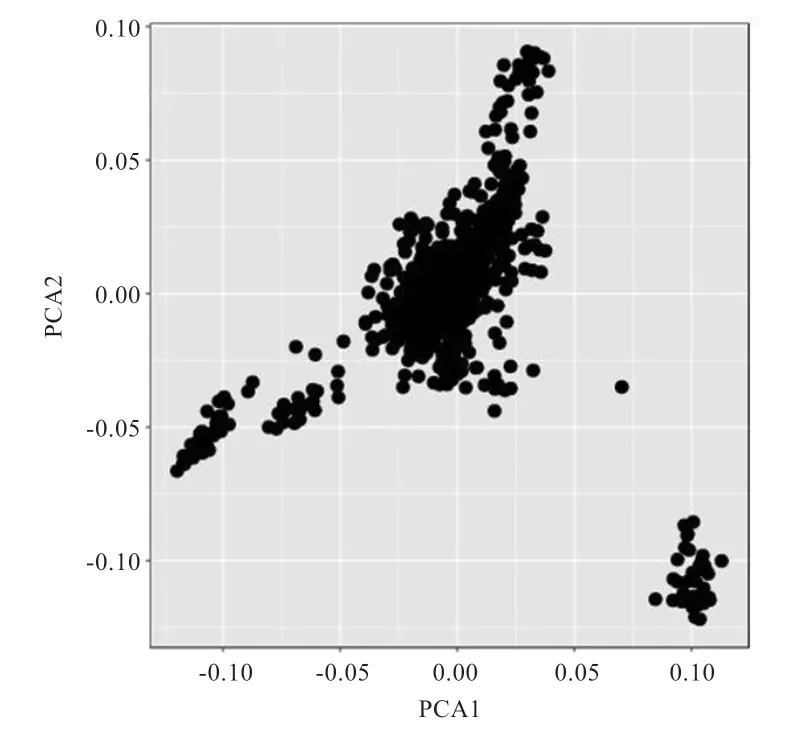
图1 群体主成分分析图
2.2 表型及基因型描述性统计
2.2.1 表型值的描述性统计 分别对840 头华西牛公牛的CW、CL 和CD 3 个性状开展单性状GWAS 与多性状GWAS 分析。表1 为各性状的原始表型描述性统计分析结果。3 个性状的表型频率分布图见图2,可以看出,各性状的表型值基本都符合正态分布。此外,对各表型值进行了表型相关分析与遗传相关分析(表2)。结果显示,胴体重、胴体长、胴体胸深3 个性状间均具有较高的表型相关(r>0.6);胴体长与胴体胸深具有中等遗传相关性(r=0.339 2),胴体重与胴体长、胴体胸深具有强遗传相关性(r>0.4)。

表1 胴体重、胴体长、胴体胸深的描述性统计
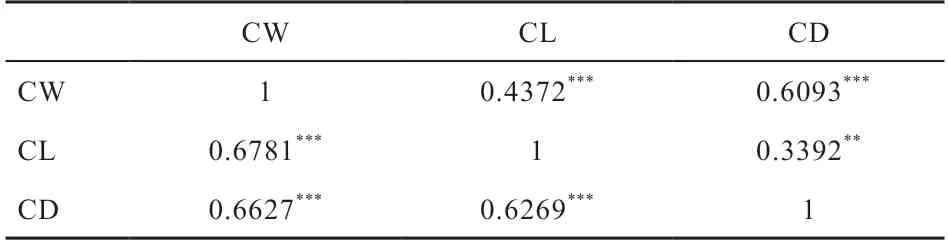
表2 性状间表型及遗传相关程度
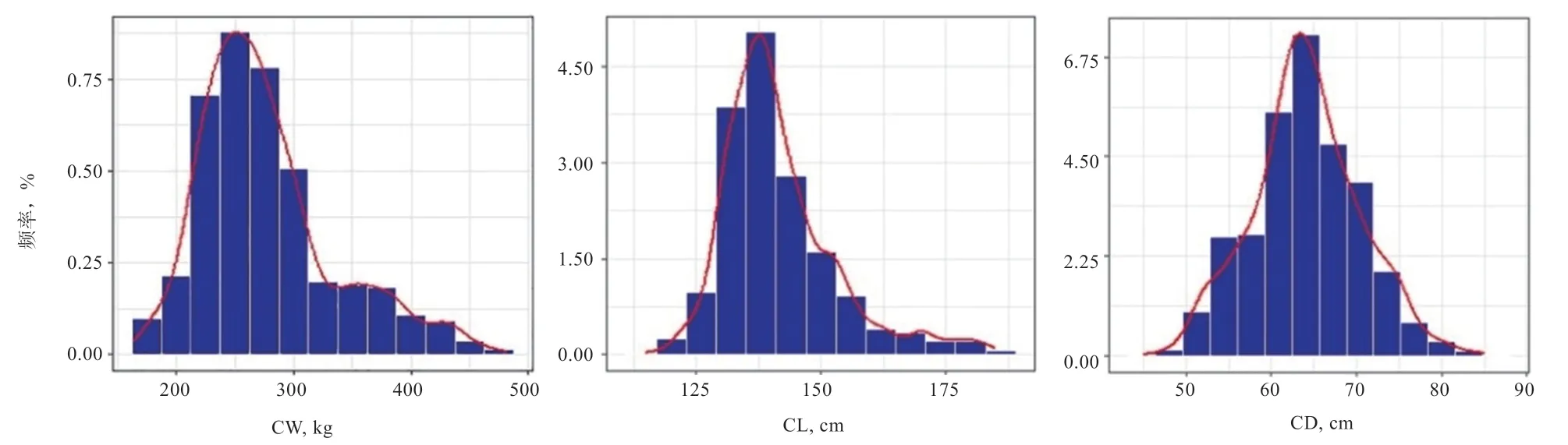
图2 CW、CL 和CD 的频率分布
2.2.2 基因型的描述性统计 按筛选原则对基因型数据进行质控后,共剩余607 198 个SNPs 标记。由图3 可知,各染色体上的SNP 位点分布较均匀,可以用于华西牛群体的GWAS 分析。
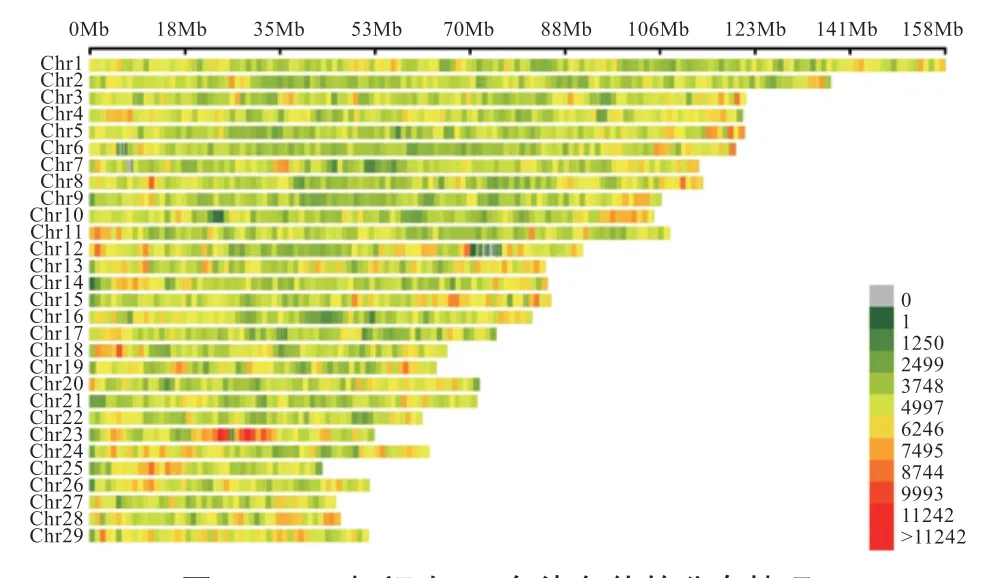
图3 SNPs 标记在29 条染色体的分布情况
2.3 GWAS 分析 使用GEMMA 软件中考虑亲缘关系的混合线性模型(LMM)对3 个性状分别做单性状GWAS 分析。曼哈顿和QQ 图如图4 所示,在CW 和CL 性状中都没有检测到显著的遗传变异位点,仅在CD 性状中发现了6 个显著的SNPs 位点,分别位于4、7、10、23、24、25 号染色体上,共注释到了5 个基因(表3)。
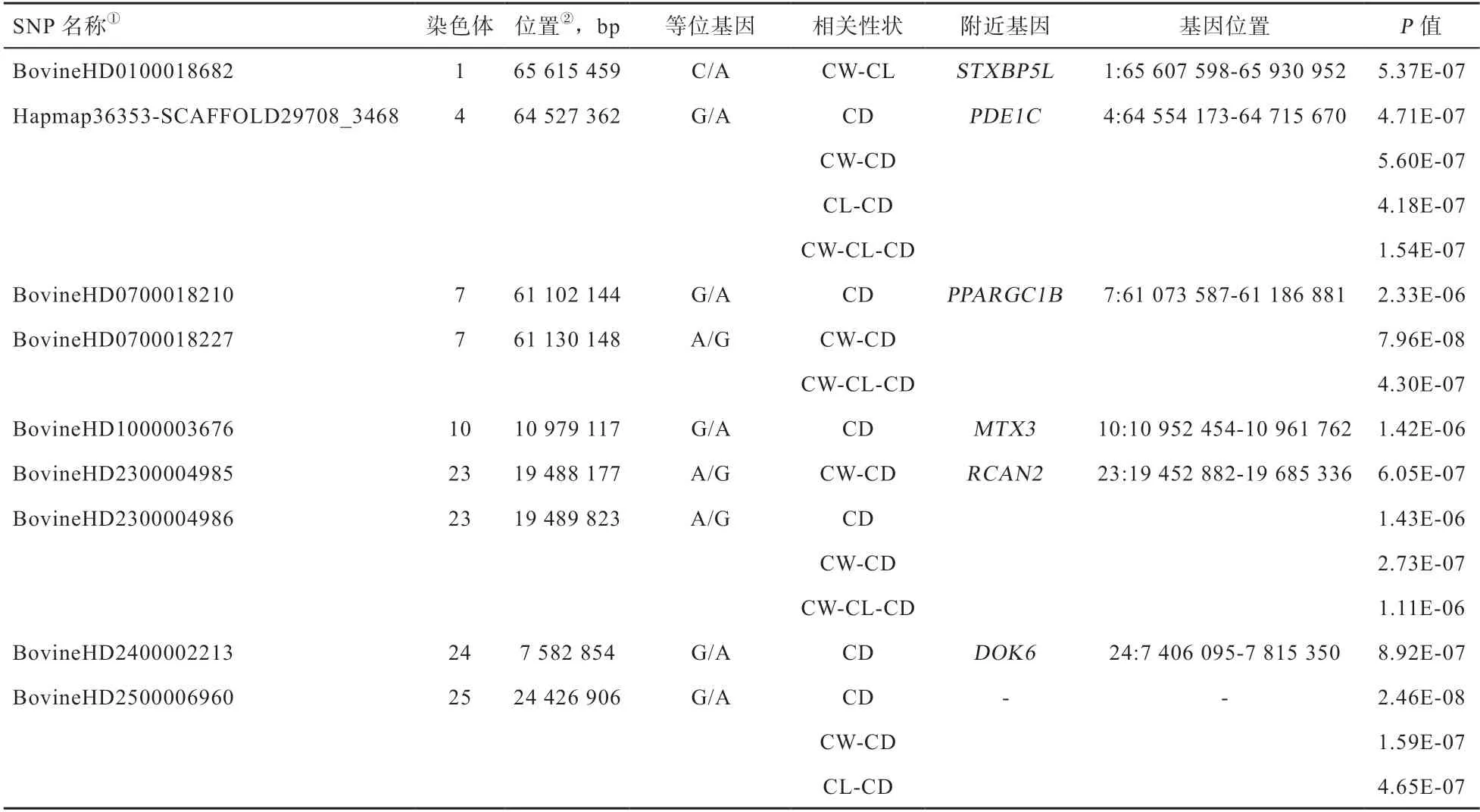
表3 单性状GWAS 和多性状GWAS 基因注释结果
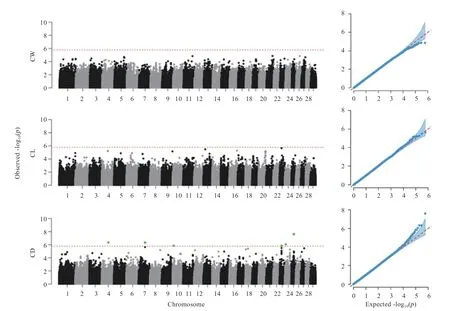
图4 单性状GWAS 曼哈顿和QQ 图
使用mvLMM 分别进行了CW-CL、CW-CD、CLCD、CW-CL-CD 4 个组合的多性状GWAS 分析(图5)。在CW-CL 组合的多性状GWAS 中,发现1 个超过染色体显著水平的SNP 位点,位于1 号染色体上,注释到了1 个基因;在CW-CD 组合的多性状GWAS 中,发现5 个超过染色体显著水平的SNPs 位点,分别位于4、7、23、25 号染色体上,共注释到3 个基因;在CL-CD组合的多性状GWAS 中,发现2 个超过染色体显著水平的SNPs 位点,分别位于4、25 号染色体上,注释到了1 个基因;在CW-CL-CD 组合的多性状GWAS 中,发现3 个超过染色体显著水平的SNPs 位点,分别位于4、7、23 号染色体上,共注释到3 个基因。合并多性状GWAS 的4 个组合的结果发现,有4 个SNPs 位点在多性状GWAS 中被重复检测到,其中Hapmap36353-SCAFFOLD29708_3468 在CW-CD、CL-CD、CW-CLCD 3 个组合中被重复检测到,BovineHD0700018227和BovineHD2300004986均在CW-CD、CW-CL-CD 2个组合中被重复检测到,BovineHD2500006960 在CLCD、CW-CD 2 个组合中被重复检测到。去除重复位点,多性状GWAS 共找到6 个一因多效的SNPs 位点。
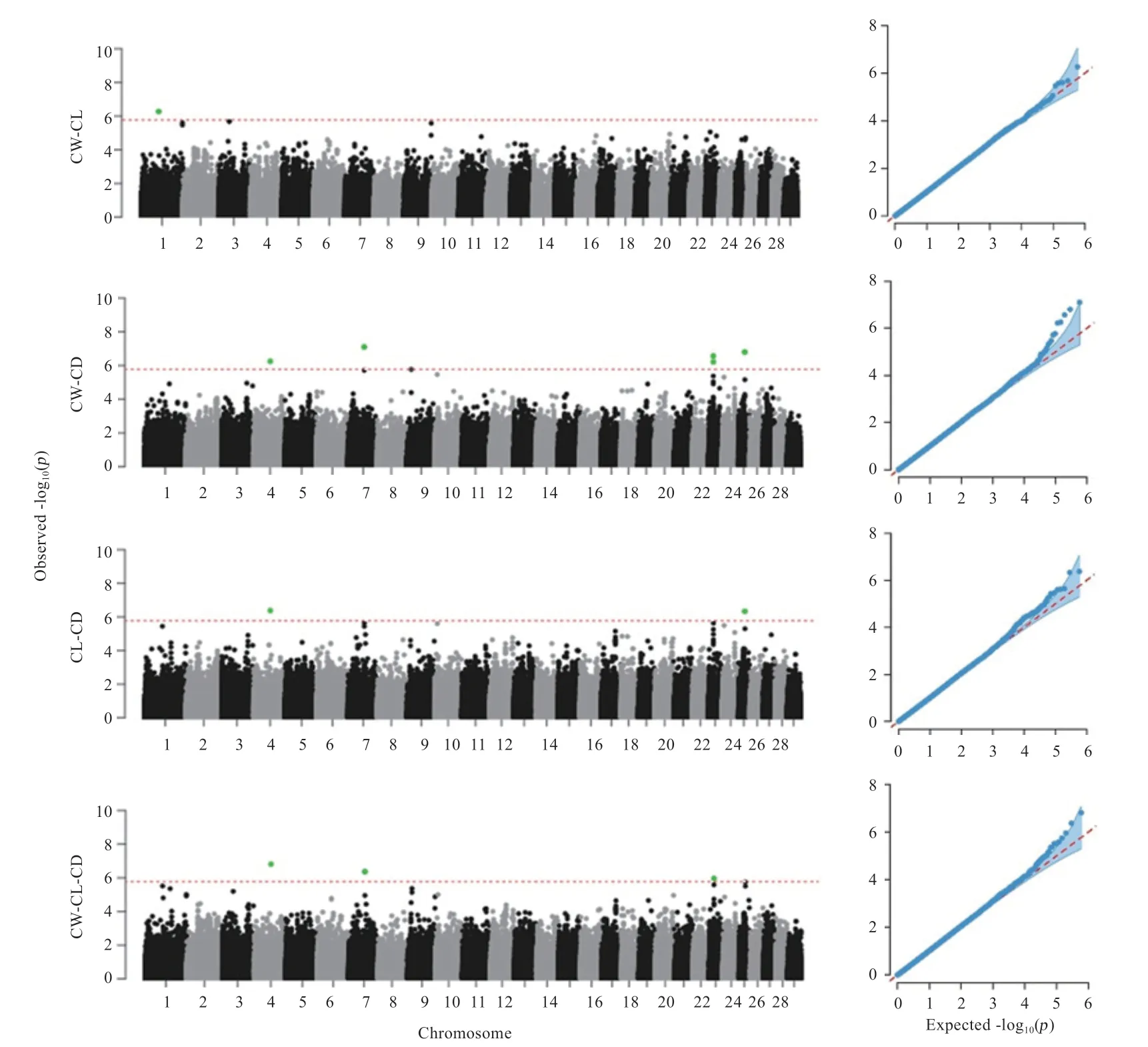
图5 多性状GWAS 曼哈顿和QQ 图
2.4 重要候选基因 对CW、CL、CD 3 个性状分别进行单性状GWAS 和不同性状之间组合的多性状GWAS结果进行汇总统计。结果得到,单性状与多性状GWAS共检测到9 个与华西牛胴体性状显著相关的SNPs 位点,根据显著SNPs 位点的物理位置,在这9 个显著SNPs 位点上下游50 kb 的区间内寻找距离最近的候选基因,共在8 个位点附近找到6 个候选基因,分别是突触结合蛋白5L(Syntaxin Binding Protein 5 Like,STXBP5L)、磷酸二酯酶1C(Phosphodiesterase 1C,PDE1C)、过氧化物酶体增殖物激活受体γ辅激活因子-1β(Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-Beta,PPARGC1B)、(Metaxin 3,MTX3)、钙调磷酸酶调节因子2(Regulator Of Calcineurin 2,RCAN2)、停靠蛋白6(Docking Protein 6,DOK6)。其中,PDE1C、PPARGC1B、RCAN23 个基因在单性状GWAS 与多性状GWAS 中被重复检测到。
由图6 可知,单性状与多性状GWAS 重复检测到的3 个显著SNPs 位点分别是Hapmap36353-SCAFFOLD29708_3468、BovineHD2300004986 和Bovine HD2500006960。其中,单性状GWAS 与多性状GWAS中CW-CD、CL-CD 组合共同检测到的BovineHD25000 06960 未注释到相关基因。此外,存在不同策略检测到的多个SNPs 位点注释到同一个基因的情况,具体为:在CD 单性状GWAS 检测到的BovineHD0700018210 与CW-CD、CW-CL-CD 2 个多性状GWAS 共同检测到的BovineHD0700018227,同时注释到基因PPARGC1B;CW-CD 多性状GWAS 检测到的BovineHD2300004985与单性状GWAS 和CW-CD、CW-CL-CD 2 个多性状GWAS 共同检测到的BovineHD2300004986,同时注释到基因RCAN2。PPARGC1B和RCAN2将作为后续重点关注的与胴体性状关联的重要候选基因。
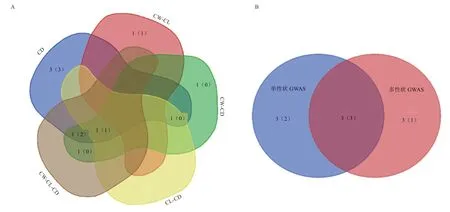
图6 单性状GWAS 与多性状GWAS 结果韦恩图
3 讨 论
在肉牛产业中,胴体性状被认为是影响肉牛生产的重要经济性状,对于提高生产效率和效益起着至关重要的作用[28-29]。然而,肉牛胴体性状的测定只能在个体屠宰后进行,这就导致胴体性状的相关表型数据收集困难且成本高昂[30]。因此,阐明胴体性状的遗传调控机制,提供可靠的分子遗传标记,可以对肉牛胴体相关性状进行早期遗传评估,从而降低育种成本,提高生产效益。
GWAS 方法已经广泛应用于动植物复杂性状的遗传解析[31-33]。本研究基于Illumina BovineHD 芯片基因分型,对华西牛CW、CL、CD 3 个胴体性状进行了关联分析,综合考虑单性状和多性状GWAS 分析得到的结果,共得到9 个显著的SNPs 位点和6 个相关基因,分别是STXBP5L、PDE1C、PPARGC1B、MTX3、RCAN2和DOK6。其中,多性状与单性状策略共同鉴定到3 个基因,分别为PDE1C、PPARGC1B和RCAN2。PDE1C基因在小鼠胚胎早期发育过程中中枢神经系统内迁移的神经元细胞中表达[34]。在人类疾病的研究中,PDE1C基因的异常会导致儿童发育迟缓[35]。PPARGC1B基因是一种调控基因转录的共激活剂,对转录因子和核受体活动、脂肪氧化、糖酵解以及能量代谢等多种生物学过程具有调控作用[36-39]。Jiang 等[40]研究了秦川牛犊牛和成年发育阶段牛脂肪组织中circRNA 的表达,发现circFUT10 在脂肪细胞增殖和分化中有重要作用,circFUT10 间接促进PPARGC1B的转录,然后调节细胞的增殖和分化,且PPARGC1B在成年牛脂肪组织中表达水平较高,说明该基因对胴体性状的表型差异可能存在重要影响。在小鼠模型中的相关研究表明,RCAN2基因对体重调节有重要作用[41-43]。RCAN2在成纤维细胞、心脏、脑、肝脏和骨骼肌中表达,能够抑制钙调磷酸酶活性,在骨骼发育和维护中有重要作用[44]。由此可见,该基因可能是影响华西牛胴体性状的重要候选基因。MTX3与DOK6是单性状GWAS 分析中鉴定到的与CD 性状相关的2 个特有基因。MTX3在蛋白质转运到线粒体这一生物过程中发挥重要作用[45-46]。DOK6能促进RET 介导的神经突生长,可能在大脑发育或维护中发挥作用[47]。Jiao 等[48]对杜洛克猪群体进行GWAS 分析,提出DOK6基因为平均日采食量、育肥期日增重、活体背膘厚3 个重要经济性状最有可能的候选基因之一。STXBP5L基因为CW-CL 组合的多性状GWAS 中单独鉴定到的基因,它在维持葡萄糖稳态[49]、囊泡运输和胞吐作用抑制[50]等过程中发挥着重要作用。
对单性状GWAS 与多性状GWAS 结果进行比较,结果发现,在相同的显著性阈值下,两者检测到的基因数目无明显差别。但是单一性状GWAS 分析中,CW、CL 性状都没有检测到显著SNPs 位点及相关基因。通过将CW 与CL、CD 两两组合进行多性状GWAS,在CW-CL、CW-CD、CL-CD、CW-CL-CD 4 个不同组合中均检测到了不同数目的显著SNPs 位点和相关基因。同时,在单性状GWAS 以及4 组多性状GWAS 中,Hapmap36353-SCAFFOLD29708_3468、BovineHD070 0018227、BovineHD2300004986、BovineHD2500006960共4 个SNPs 位点,以及PDE1C、PPARGC1B、RCAN2共3 个基因被重复检测到,推测认为这些位点及基因极大可能是潜在的一因多效位点和一因多效基因。总体而言,多性状GWAS 可以提高对位点的检测效率和准确性,这与高进等[51]、Bolormaa 等[16]的研究结果相一致。在实际育种当中,CW、CL 和CD 的表型通常是同时被选择的,由于多性状GWAS 分析检测到的SNPs 位点具有一因多效的优越性,因此综合考虑多性状GWAS 分析在生物学和实际应用层面更有意义。
4 结 论
本研究基于Illumina BovineHD 高密度基因分型结果,利用840 头华西牛胴体重、胴体长和胴体胸深表型数据,采用单变量和多变量线性混合模型方法进行单性状和多性状的GWAS 分析,共检测到9 个显著关联的SNPs 位点,注释到STXBP5L、PDE1C、PPARGC1B、MTX3、RCAN2、DOK6共6个候选基因,其中PPARGC1B、RCAN2被不同方法检测到的多个SNPs 位点同时注释到,且参与体重调节、脂肪细胞增殖和分化等过程,推测其可能是影响胴体性状的重要候选基因。
猜你喜欢
能源研究与信息(2022年3期)2022-10-29
中国现代医生(2022年21期)2022-08-22
中国饲料(2021年17期)2021-11-02
天津医科大学学报(2021年1期)2021-01-26
医药前沿(2020年20期)2020-11-10
三农资讯半月报(2020年2期)2020-03-09
世纪人物(2015年3期)2015-12-28
文史博览·文史(2015年8期)2015-08-18
世纪人物(2015年1期)2015-01-28
食品工业科技(2014年5期)2014-03-11
