
基于整合方法分析茶树响应病原真菌胁迫的共有模式
2023-03-09 02:13连玲丽陈强周颖付婧李婉莹魏日凤刘伟
热带亚热带植物学报 2023年1期
连玲丽, 陈强, 周颖, 付婧, 李婉莹, 魏日凤, 刘伟
基于整合方法分析茶树响应病原真菌胁迫的共有模式
连玲丽1a, 陈强1b, 周颖1a, 付婧1a, 李婉莹1a, 魏日凤1b, 刘伟2*
(1. 福建农林大学, a. 生命科学学院; b. 园艺学院, 福州 350002; 2. 宁德师范学院, 福建 宁德 352100)
为探讨茶树()对病菌胁迫的共有响应模式和抗病机制,运用生物信息学方法对多组RNA-seq数据进行提取、整合及功能富集,结合多种工具和数据库资源对主要调控分子及蛋白互作模块加以分析。结果表明,病原真菌胁迫下,茶树有较多细胞色素P450家族成员表达显著上调;类固醇和激素的代谢过程、苯丙烷合成途径被激活,有丝分裂细胞周期调控、DNA甲基化等生物过程及光合作用途径受到抑制;主要调控分子如转录因子和、激酶和等以上调为主。差异表达的蛋白互作模块分析表明,有丝分裂周期调控、基于微管运动、淀粉和蔗糖代谢、细胞壁多糖合成、光合作用、类黄酮代谢模块明显下调,木质素合成和萜类生物合成模块上调;且模块之间可能存在互作。病菌胁迫激活的木质素和萜类合成途径的关键基因包括阿魏酸-5-羟基化酶基因、过氧化物酶基因和萜类合成酶基因等。细胞色素基因可能在病菌胁迫中起关键作用,增强木质素和萜类物质的合成、削弱光合作用可能是茶树响应真菌胁迫的核心模式。
茶树; 病原真菌; 病菌胁迫;整合分析
茶树()作为一种重要经济植物,在我国东部和南部各大茶区广泛种植,近年来种植范围更是向北延伸至河北、向西进入西藏[1]。然而, 随着全球气候变化加剧,恶劣天气使病虫害发生呈现加重趋势,给茶树种植业带来严重损失。其中同样由炭疽病菌属(spp.)引起的茶叶炭疽病和云纹叶枯病作为茶园中重要的叶部病害,更是对茶叶的产量和品质造成直接影响[2]。如何提高茶树对病菌胁迫的抗性是生产中亟待解决的关键问题。因此,本研究基于整合分析方法探究茶树响应病菌胁迫的共有途径或基因,以期为明确茶树抗病机制、提高茶树抗病性等深入的研究提供参考。
伴随着茶树中国种基因组草图和精细测序数据的公布[3–4],人们得以结合组学技术更深入地探讨茶树在病菌胁迫下的响应机制,Wang等[5]报道茶树抗感品种对炭疽病菌()的响应差异体现在苯丙烷和类黄酮等物质合成的差别;Lu等[6]的研究表明茶树与炭疽病菌的互作主要与胼胝质的沉积、多种不同激素的信号转导有关;茶树响应叶斑病菌(、)侵染时,淀粉与蔗糖代谢、苯丙烷生物合成、植物激素信号、类黄酮生物合成等代谢途径呈现显著增强[7–8]。这从不同角度展现了茶树响应病菌的可能机制,但未能提供茶树抵抗病菌的共性信息。另一方面,茶树在生产中常出现不同病菌复合侵染的现象,因而突破以往仅针对单一病菌胁迫的研究范式、探讨茶树对多种病菌胁迫响应的共性机制, 具有重要的实践意义。
整合分析方法作为一类统计方法,对围绕同一问题的多个独立研究进行综合分析,既能充分利用多项研究的信息,也能使我们对目标问题有更深的理解,因而被广泛应用于医学、农学等领域[9],近年来也越来越多地被用于组学数据的分析。如Ashrafi-Dehkordi等[10]对番茄()在不同胁迫下的转录响应进行整合分析,检测得到响应不同胁迫的通用基因,也发现了54个新的差异表达基因;Panahi等[11]通过跨物种的整合分析确定了重要的盐胁迫响应途径等。目前,利用整合分析方法挖掘茶树转录组数据资源的相关研究仍未见报道,我们以茶树生产中常见的不同病菌复合胁迫问题为切入点,结合生物信息学方法开展相关研究。通过整合茶树响应不同病菌胁迫的组学数据, 克服单一研究可能存在的局限性,确定整合方法在茶树相关研究领域应用的可行性;在获取、整合差异表达基因数据集的基础上,结合功能富集和网络模块等分析方法明确共有核心响应分子及途径,为后续的关键分子功能研究及育种应用奠定理论和实践基础。
1 材料和方法
1.1 数据获取
以“([Title]) AND infected[Text Word]”为关键词在NCBI-SRA数据库检索,再通过过滤词“Source:RNA”、“Library Layout:paired”和“Platform:Illumina”进行过滤,获取含有对照和病菌胁迫的茶树RNA-seq数据;同时,以“(Tea Plant [Title]) OR ([Title]) AND (resistance [Abstract]) AND (transcriptome[Abstract])”为检索词搜索NCBI PubMed数据库,手工选择搜索结果中与病菌侵染茶树有关的文献,并从文献中查找数据源。
通过数据库检索确定了PRJNA396805、PRJNA 528172、PRJNA637492和PRJNA564655等4项同时包含对照和病菌处理的茶树转录组研究[5,7–8,12],经样本间PCA分析,去除样本分组效果不佳的部分研究,最终保留了3项研究(表1)共计24份表达数据用于整合分析。
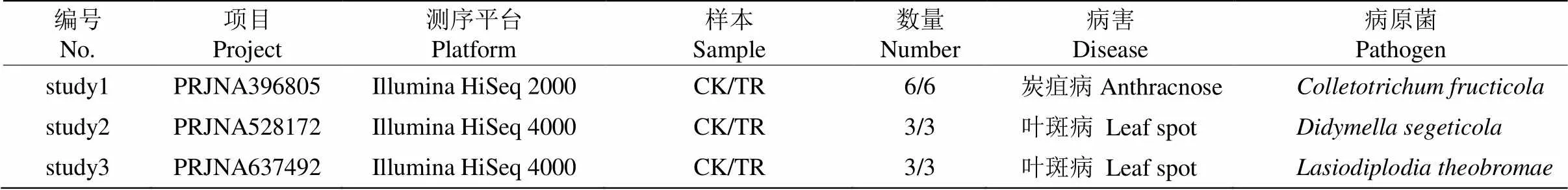
表1 用于整合分析的转录组数据集
CK: 对照; TR: 接种病原。
CK: Control; TR: Inoculation of pathogen.
1.2 数据处理
采用FastQC v 0.11.8对每个样本进行质量检测,再由Trimmomatic 0.39去除接头和低质量区域,获得清理后的读段;继而采用HISAT2将清理后的读段匹配至茶树“舒茶早”基因组,采用feature Counts对匹配结果进行计数;最后使用DESeq2 (v 1.32.0)对各项研究进行差异表达分析,视Log2Fold Change(Log2FC)绝对值>1、校正后值<0.05的基因为具有统计意义的差异表达基因(DEGs)。
1.3 整合分析鉴定差异基因
基于值合并法,通过metaRNASeq[13]的Fisher结合概率法对独立研究的差异表达基因(individual- DEGs)进行整合,其中采用Benjamini-Hochberg错误发现率对原始值进行校正,取校正后值<0.05且表达趋向一致的基因作为整合分析后的差异表达基因(meta-DEGs);继而构建individual-DEGs和meta-DEGs的韦恩图,并提取单项研究与整合分析的共有差异表达基因(common-DEGs)用于后续功能分析。
1.4 功能预测与分析
为确定茶树响应病菌胁迫的关键生物过程或途径,利用eggNOG在线工具(http://eggnog5.embl.de/)重新注释茶树基因组蛋白序列的GO和KEGG信息,再由clusterProfiler对meta-DEGs进行功能富集分析,保留其中校正值<0.05的结果。借助iTAK v 1.6 (http://itak.feilab.net/cgi-bin/itak/online_itak. cgi)鉴定meta-DEGs中的转录因子和蛋白激酶,取值<1E-5的结果;同时基于miRNA的数据库资源(plant microRNA database, http://bioinformatics.cau. edu.cn/PMRD/),通过TAPIR靶标预测工具(http://bio informatics.psb.ugent.be/webtools/tapir/)确定可能受miRNA调控的差异表达基因。借助STRING数据库(http://string-db.org)基于高可信度互作记录绘制meta-DEGs的蛋白互作网络,以确定茶树响应病菌胁迫的关键模块。
2 结果和分析
2.1 整合分析结果
将各项研究的基因表达信息分别匹配到茶树“舒茶早”基因组的33 932个编码基因上,并经差异分析和整合处理,获得3个独立差异基因集(individual- DEGs)和1个整合差异基因集(meta-DEGs)。其中, 独立差异基因集的基因数量差别较大,尤其是同样与叶斑病胁迫相关的study2和study3,分别涉及2 515和10 699个差异基因。通过值合并法整合得到的meta-DEGs包括3 335个差异基因,涉及2 093个上调表达基因和1 242个下调表达基因。从individual- DEGs与meta-DEGs结果的交叠情况来看(图1),独立差异基因集内有23.76%~40.83%的基因仍保留于整合结果;而整合结果也有近40%的差异基因与至少2个独立研究的表达趋势一致,表明整合结果在较大程度上包含了多项研究的信息。
进一步了解4个基因集的共有差异基因和整合分析的特有差异基因,可知共有差异基因集(common- DEGs)包含的305个基因中有约30%的基因编码未知蛋白,40%的基因编码酶类物质,包括激酶、甲基化转移酶和维生素C氧化酶等;整合分析获得192个新的差异基因,除了多数编码未知蛋白的基因之外,还有部分编码受体样蛋白、激酶及抗病蛋白的基因,总体上表达量的变化倍数为1~2。
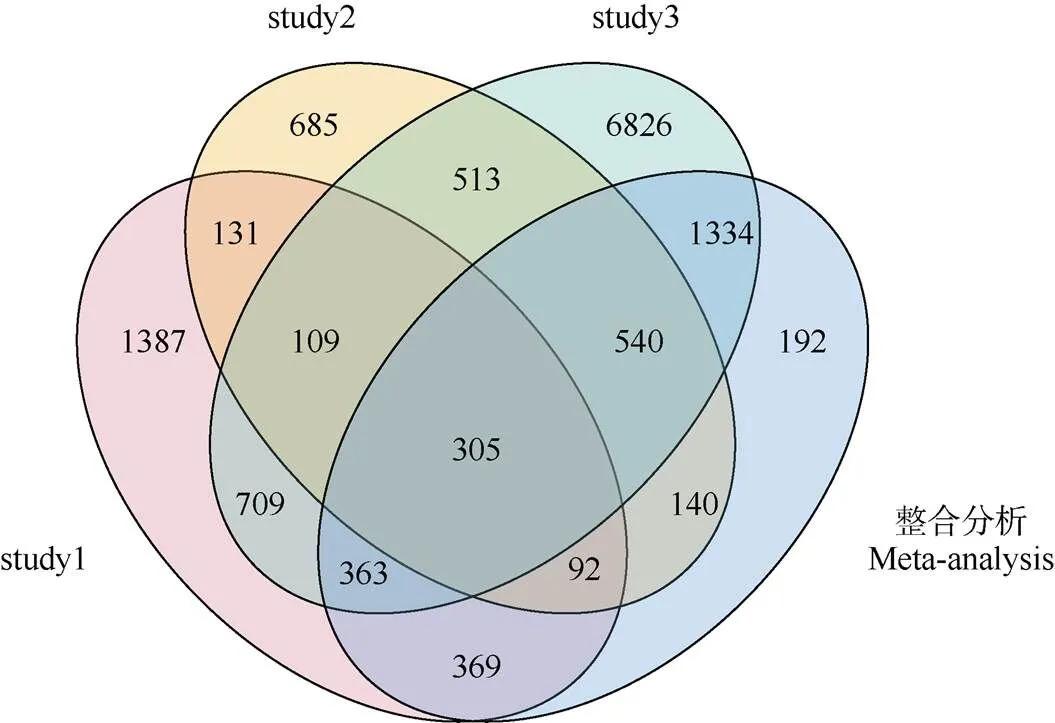
图1 独立研究与整合分析的差异表达基因集的关系
以差异倍数的绝对值为主要排序依据,取meta-DEGs中表达量变化倍数最大且FDR值较小的前30个DEGs列于表2。编码细胞色素P450的基因出现次数最多(8/30)且均上调表达,编码漆酶、病程相关蛋白、激酶和几丁质酶等基因的差异表达水平也较高, 同样呈现上调表达;这些基因中有不少是独立研究及整合分析的共有基因,如细胞色素P450基因(TEA010837)、漆酶基因(TEA020596)等,表明它们更可能是茶树响应不同病菌胁迫的关键基因。
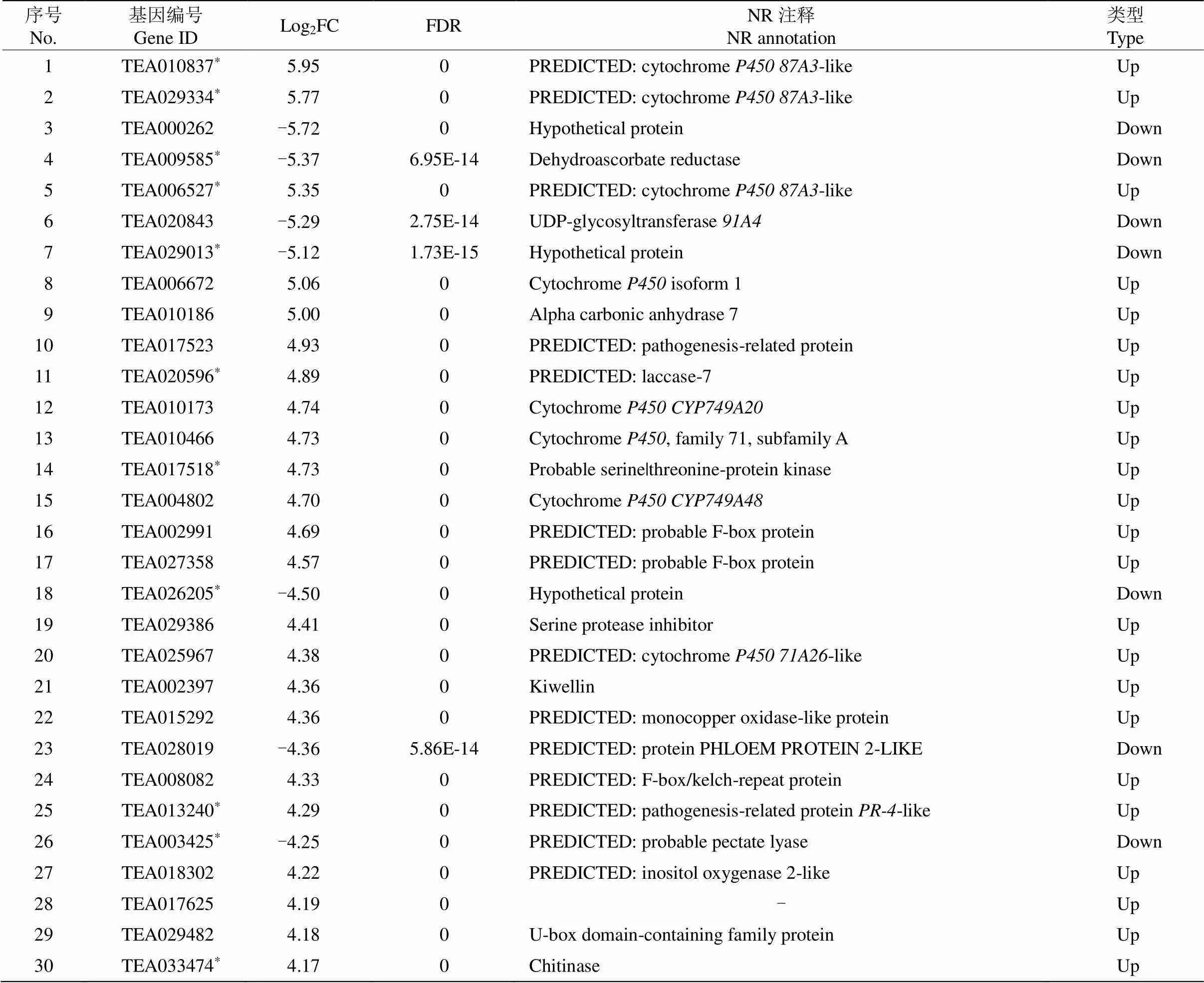
表2 整合分析鉴定的前30个差异表达基因
*: 共有DEGs。下同
*: Common-DEGs. The same below
2.2 整合结果的功能富集分析
对整合分析获取的上调和下调差异表达基因分别进行GO生物过程和KEGG代谢途径的富集分析(表3)。上调表达基因的富集结果中,GO富集分析确定了病菌胁迫下茶树的类固醇代谢、激素代谢、有毒物质响应、苯丙烷代谢等生物过程增强, KEGG分析同样富集到油菜素类固醇合成、细胞分裂素(玉米素)合成、苯丙烷生物合成等相似的途径; 而下调表达基因的富集结果则表明,病菌胁迫下茶树的细胞壁组织生成、有丝分裂细胞周期调节、DNA复制、微管解聚等生物过程减弱,黄酮类和萜类等物质的合成、淀粉与蔗糖代谢、光合作用等相关代谢途径受到抑制。

表3 整合分析后差异表达基因的功能富集
以上调为主的苯丙烷合成途径和显著下调的光合作用途径也分别被common-DEGs的上调和下调子集所富集,其中富集到苯丙烷合成途径的关键基因有编码阿魏酸-5-羟基化酶的基因(TEA032005和TEA000057)、过氧化物酶基因(TEA001789、TEA028696)、糖苷水解酶基因(TEA004253)和-葡萄糖苷酶基因(TEA001710);富集到光合作用途径的基因有编码光系统I的和基因(TEA017139、TEA001351、TEA002548和TEA033829),表明这2个代谢途径在茶树响应病菌胁迫中起重要作用。
2.3 响应病菌胁迫的转录因子、激酶和miRNA的鉴定
转录因子(TFs)和激酶(PKs)在植物响应生物胁迫中发挥重要作用。通过将meta-DEGs比对到转录因子数据库中,获得207个转录因子,归属于38个家族,其中、/、、、和等家族均有10多个成员发生显著表达变化。差异表达基因中,上调表达的数量明显少于下调表达的(63 up/174 down),表明病菌胁迫下转录因子的表达普遍受到抑制。但也有部分转录因子的表达以激活为主,如家族(13up/1down)和家族(9 up/7 down);还有部分转录因子的表达虽以抑制为主,但仍有一定数量的成员被激活,如家族(5 up/20 down)和(4 up/21 down), 表明这些转录因子相较于其他转录因子在茶树响应病菌胁迫中发挥更为重要的作用。

图2 差异表达基因中转录因子家族的分布
利用iTAK工具在meta-DEGs中鉴定得到308个蛋白激酶,涉及AGC、CAMK、CMGC、Group-Pl、RLK、STE和TKL等多种激酶家族,其中受体蛋白激酶RLK的成员数量最多(269个)、其次是钙调激酶CAMK(6个)。这2个家族的成员均以上调表达为主,分别有160和4个成员表达量显著上升; 其他家族成员则多数是下调表达。在差异表达的家族中,亚家族、、、和的绝大多数成员呈现上调表达;相反地,亚家族、等的成员则多为下调表达,表明的亚家族可能在茶树响应病菌胁迫中扮演不同角色。
miRNA参与基因表达的调控。通过miRNA-靶标基因关系的预测,可知病菌胁迫下78个下调表达的茶树基因可能受到53个miRNA (归属于23个miRNA家族)的调控。从表4可见,调控的基因数量最多(34个),这些基因参与有丝分裂细胞周期过程、植物类细胞壁组织发生等生物过程,其次和分别抑制漆酶和生长调节因子的表达,表明这些miRNA可能是重要的胁迫相关调控因子。此外,和等保守家族也参与茶树的胁迫响应,调控的基因负责编码转录因子、K+外排逆向蛋白和同源盒亮氨酸拉链蛋白。
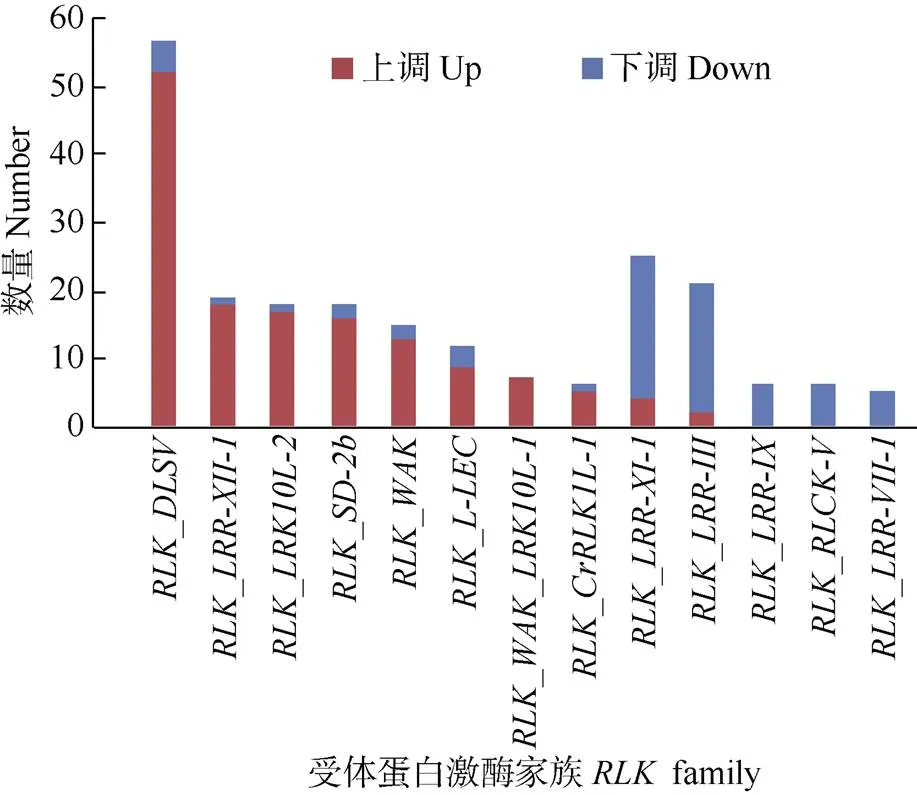
图3 差异表达基因中蛋白激酶RLK亚家族的分布
2.4 蛋白互作网络分析
将meta-DEGs的基因映射到拟南芥蛋白互作关系中,预测病菌胁迫下茶树差异表达基因之间的可能互作模式。由构建的蛋白互作网络中提取核心区域作图,并分析其中的主要功能模块(图4),获得的8个功能模块分别是有丝分裂周期调控(M1)、基于微管运动(M2)、淀粉与蔗糖代谢(M3)、细胞壁多糖的合成(M4)、光合作用(M5)、类黄酮和类固醇合成代谢(M6)、木质素合成(M7)、萜类合成(M8)。其中模块M1~M5的节点对应基因几乎都呈现下调表达, 模块M6中与黄酮醇的合成、花青素合成有关的节点对应基因(、、、)下调表达,与油菜素甾醇合成有关的节点(细胞色素P450)对应基因(TEA015379、TEA015397)上调表达;M7和M8的节点均以上调表达为主,分别与木质素的合成与代谢、萜类骨架合成有关。
从图4可见,模块之间相对独立,但彼此又存在不同程度的交互作用,其中模块M1和M2、M6和M7间的关系紧密,有较多跨模块的互作,如模块M6的黄酮醇合酶FLS (TEA010328)、二氢黄酮醇还原酶DFR (TEA024762)均与模块M7的莽草酸羟基肉桂酸酰转移酶HCT (TEA022314、TEA011691)互作;其他模块间则通过单对蛋白节点的互作建立联系,如模块M5的早期光诱导蛋白ELIP1 (TEA009875)与模块M6的DFR互作、模块M5的PSII色素结合蛋白NPQ4 (TEA019348)与模块M1的蛋白激酶STN8 (TEA020777)互作、模块M5的核酮糖-1,5-二磷酸羧化酶/加氧酶Rubisco (TEA004730)与模块3的26S蛋白酶体非ATP酶调节亚基RPN (TEA000905)互作等。这些参与模块间互作的桥梁分子可能在模块的协同响应病菌胁迫中发挥作用。

表4 预测的miRNA及其靶向的下调基因
*: 受2个miRNA家族调控; **: 功能未知, 共计21个。
*: Regulated by two miRNA families; **: Unknown function, 21 in total.
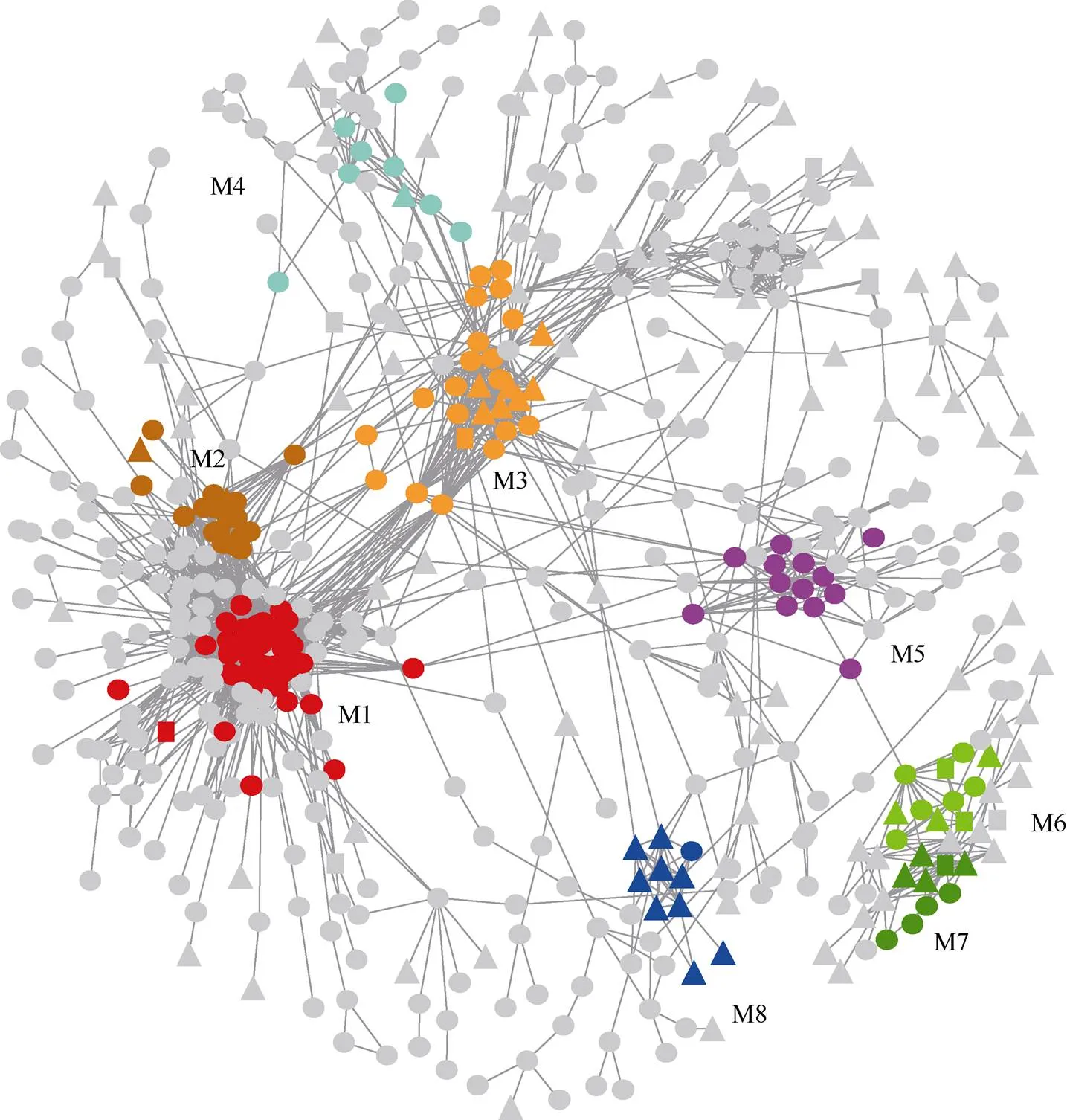
图4 茶树响应病菌胁迫的核心网络的模块分布。三角形: 表达上调; 圆形: 表达下调; 方形: 表达上下调兼有。M1:有丝分裂周期调控; M2: 基于微管运动; M3: 淀粉与蔗糖代谢; M4: 细胞壁多糖合成; M5: 光合作用; M6: 类黄酮和类固醇合成; M7: 木质素合成; M8: 萜类合成。
进一步提取以上调表达为主的模块M7和M8的互作关系,并将其映射到代谢途径中。从图5可见,模块M7中木质素合成相关酶类的基因(、、、、和)及萜类合成相关的催化酶基因(、、、和) 几乎都是上调表达,其中(TEA000057和TEA 032005)、(TEA028696)和(TEA021767)不仅在不同病菌胁迫下均呈现上调表达,且上调程度最为显著,表明它们是这些代谢途径中更为重要的分子。从代谢途径的蛋白互作情况来看,由l-苯丙氨酸到木质素的代谢流内存在的互作关系主要发生在相邻酶分子之间,也有个别酶分子与非相邻酶分子互作,如PAL与COMT、CcoAOMT、CAD等的互作,表明这类酶可能在整个代谢调控中占据重要地位。
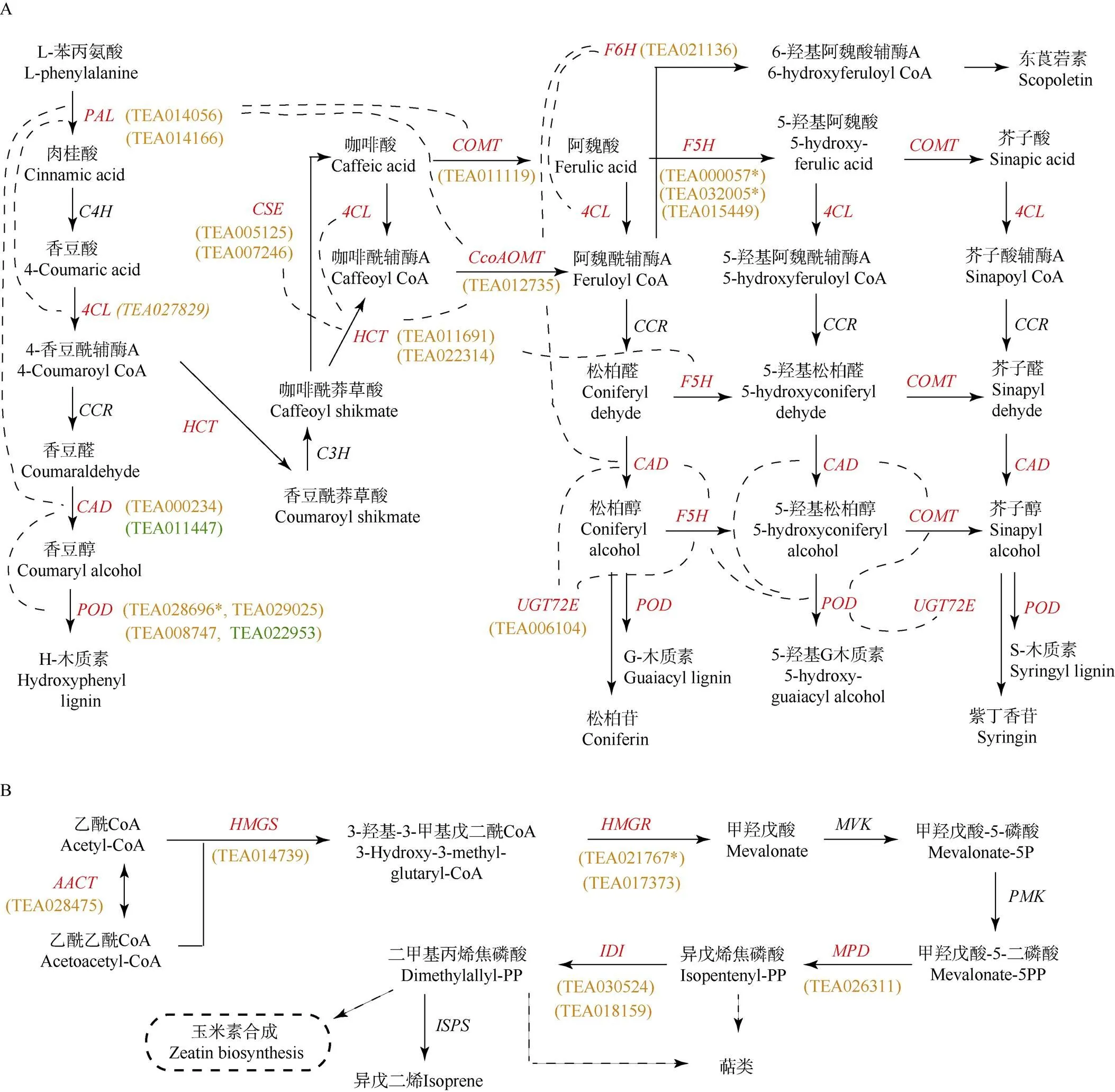
图5 木质素合成模块(A)和萜类的甲羟戊酸合成模块(B)及蛋白互作。红色: 差异表达的酶基因; 黄色: 上调表达基因; 绿色: 下调表达基因; *: 共有基因; 虚线: 互作关系。
3 结论和讨论
测序技术的发展和转录组研究数量的增多,使得我们有可能基于多份组学数据获取共有的表达信息,以减小单次研究可能存在的分析偏颇。本研究首次尝试对多份茶树响应病菌胁迫的组学数据进行整合分析,以挖掘共有信息。在整合得到的3 335个差异表达基因(DEGs)中,显著下调的DEGs居多(62.76%),但在表达倍数变化最大的前30个DEGs中,显著上调表达的更多(76.67%),表明茶树在遭受病菌侵染时可能从整体上降低代谢水平, 并将有限的能量用于激活特定的生物分子。这些被激活的分子中,存在多个细胞色素P450家族成员, 还包括碳酸酐酶(TEA010186)、病程相关蛋白(TEA017523)和漆酶(TEA020596)等抗病相关基因, 它们的功能在以往研究中虽有报道[14–16],但在茶树响应病菌胁迫中的作用仍有待明确。
为了解茶树在病菌胁迫下基因调控的有关信息,对差异表达的转录因子、激酶和miRNA进行分析。结果表明,病菌胁迫下来自38个家族的207个转录因子的表达发生明显变化,其中和转录因子的成员数目最多,以下调表达为主;和转录因子成员数目次之,以上调表达为主,表明这些家族可能参与茶树响应真菌胁迫的负向或正向调控。在这4类转录因子中,茶树家族的功能研究较多,不仅通过qRT-PCR技术验证了多个茶树基因在植株接种炭疽病菌后呈现表达量的显著上调[17]、还利用基因沉默试验证明缺失基因的植株对病原真菌更为敏感[18];相比而言,其他3类茶树转录因子的功能研究较少。尽管如此,其他物种的相关研究证实了它们同样在植物响应病菌胁迫中发挥重要功能,转录因子对水稻()的稻瘟病抗性、小麦()的白粉病抗性、番茄的灰霉病抗性及马铃薯()的晚疫病抗性等均具有正向调控作用[19];烟草()的基因沉默使植株对晚疫病菌更加敏感[20],辣椒()的基因的过表达增强植株对病原真菌的抗性[21];棉花()的转录因子正向调控植株对黄萎病菌的抗性[22]等。因此,这些转录因子很可能也在茶树抗病响应的基因调控中扮演重要角色。对茶树在病菌胁迫下差异表达的蛋白激酶(PKs)进行分析,结果检测到308个PKs,其中绝大部分来自家族,其次是家族。在其他植物全基因组水平的PKs分析中,这2个家族的数量也较多, 两者均参与植物的逆境胁迫响应,葡萄()的CAMK家族成员在植株响应非生物胁迫中发挥作用[23];豇豆()接种病毒后,亚家族和家族上调表达的成员数量明显多于其他家族[24]。在茶树中,家族成员参与植株对寒害胁迫的响应[25],至于亚家族、家族在茶树响应病菌胁迫中的作用则未见报道。因此,开展相关的后续研究有助于解析两者在茶树对抗真菌病害中的具体功能。对茶树在病菌胁迫下差异表达的miRNA进行预测,得到来自23个家族的53个miRNA,其中靶标基因数量或成员数量较多的miRNA来自、、、等家族,它们的靶向基因主要是转录因子。Jeyaraj等[26]预测、、和在茶树受炭疽菌侵染后发生显著的表达变化,且可能靶向调控转录因子(如、)和抗氧化酶基因(如、)的表达;更多的研究表明,和家族成员参与植物对不同病原真菌的防御响应,如黄瓜()对霜霉病菌侵染[27]、水稻对稻瘟菌侵染的响应[28]等。因此,这2个miRNA家族可能在茶树抗病响应中发挥相似的作用。
为进一步明确茶树在病菌胁迫下发生变化的生物过程及代谢模块,对整合差异表达基因(meta- DEGs)进行功能富集分析。结果表明,病菌胁迫下类固醇代谢、激素代谢和苯丙烷代谢等显著增强,这与单项研究的结果基本一致。结合KEGG富集分析结果,相关代谢被进一步细化至苯丙烷、油菜素内酯和玉米素的合成途径,其中苯丙烷合成途径也在油茶响应炭疽病菌胁迫时被激活[29],表明该合成途径可能是茶树响应病菌的核心途径。结合蛋白互作网络模块来看,与苯丙烷代谢密切相关的2个分支途径中,木质素合成模块内合成相关酶对应的基因明显上调表达,类黄酮合成模块内与花青素、黄酮醇合成相关的酶类基本呈现下调表达,其中前者的代谢与植物抗性相关,后者的代谢则与植物品质有关[30],表明茶树在响应病菌胁迫时可能以损失品质为代价来加强自身的抗病性。类似地,甲羟戊酸(油菜素内酯及玉米素的前体物质)的合成途径也在病菌胁迫时被激活。在月季()抗灰霉病菌的转录组学分析中,与油菜素内酯和乙烯相关的基因表达水平都明显高于水杨酸和茉莉酸的,且油菜素内酯在胁迫响应中发挥主导作用[31]。由此可见,木质素合成途径、油菜素内酯合成途径可能是茶树防御病菌胁迫的重要代谢途径。
除上调表达的代谢途径外,整合差异表达基因的功能富集分析还表明,茶树在病菌胁迫下受到抑制的代谢途径,包括基于微管的过程、有丝分裂细胞周期及其调节、淀粉和蔗糖代谢、光合作用等途径的显著下调。其中,光合作用不仅在其他植物响应病原真菌胁迫中发生明显的下调变化[32],也在植物响应寒冷、高温、病原细菌等多种胁迫中受到抑制[33],表明光合作用削弱可能是植物逆境响应的一般模式。编码与光合作用相关的光系统I组成蛋白psaA和psaB、光系统II的PSBS蛋白和PSBO蛋白、ATP合酶亚基等的基因均呈现下调表达,尽管这些光系统组分在植物抗真菌病害中的作用仍鲜见报道,但已有研究[34]表明,病原细菌效应蛋白及病毒蛋白均能靶向光系统组分蛋白以破坏光系统, 也有研究表明[35–36]增强光系统组分的表达或活性能提高植株对病原细菌或花叶病毒病的抗性,这些研究表明光合作用系统在植物-病原互作中占据重要位置。除此之外,结合蛋白互作网络模块分析的结果,光合作用模块与有丝分裂周期调控模块、淀粉与蔗糖代谢模块及次生代谢物模块之间可能存在模块间的互作,这与“光合作用可能是调节及平衡基础代谢与防御相关代谢的重要枢纽[34]”的观点一致。因此,明确茶树光合系统在调控防御响应中发挥的作用将有助于加速抗病机制、品种选育等研究的进程。
结合蛋白互作预测方法了解茶树在病菌胁迫下可能存在的蛋白互作信息,着重对以上调表达为主的木质素合成模块和萜类合成模块的互作进行分析。结果表明,病菌胁迫下茶树的这2个合成途径中相关合成酶之间存在较多互作。在木质素合成途径中,预测的PAL与4CL、COMT、CcoAOMT的互作,HCT与4CL、CSE的互作,以及CAD与CcoAOMT的互作均在毛果杨()木质素生物合成酶的互作试验中得到验证,且多数为瞬时互作[37],其中HCT与4CL能形成蛋白复合体促进木质素的合成[38];合成途径中POD与CAD、UGT72E、F5H的互作则仅出现于其他相似的预测研究[39]中,尚未有试验验证的报道。在萜类合成途径中,合成相关酶AACT、HMGS、HMGR、MPD和IDI之间可能存在互作,其中AACT与HMGS的互作在古细菌和真细菌中得到证实[40],而HMGR和HMGS、MPD的互作则出现于葡萄的HMGR蛋白互作预测结果[41]中。这些研究暗示了合成途径中酶的互作既有保证正常代谢的稳定互作,也有应对环境改变而产生的瞬时互作,同时酶的互作可能不同程度地调控代谢终产物的合成量,进而影响植物表型。因而,探明茶树中与病菌防御相关代谢途径的完整互作网络及逆境胁迫下的动态互作网络, 将有助于从分子层面揭示茶树的抗病分子机制。
综上,茶树在响应不同病原真菌胁迫时均增加木质素合成、降低光合作用,且更倾向于通过甲羟戊酸途径合成萜类物质或激素。、、和等转录因子为参与胁迫响应的重要分子,这为后续的关键途径与分子的功能研究、茶树的抗病品种选育提供参考信息。
[1] TANG J X, WANG P J, E Y H, et al. Climatic suitability zoning of tea planting in China’s mainland [J]. J Appl Meteor Sci, 2021, 32(4): 397– 407. [唐俊贤, 王培娟, 俄有浩, 等. 中国大陆茶树种植气候适宜性区划 [J]. 应用气象学报, 2021, 32(4): 397–407.]
[2] CAI X M, LUO Z X, BIAN L, et al. Tea pest prevention and control progress during the 13th five-year plan period and development direction in the 14th five-year plan period [J]. China Tea, 2021, 43(9): 66–73. [蔡晓明, 罗宗秀, 边磊, 等. 茶园绿色防控“十三五”进展及“十四五”发展方向 [J]. 中国茶叶, 2021, 43(9): 66–73. doi: 10.3969/ j.issn.1000-3150.2021.09.010.]
[3] WEI C L, YANG H, WANG S B, et al. Draft genome sequence ofvar.provides insights into the evolution of the tea genome and tea quality [J]. Proc Natl Acad Sci USA, 2018, 115(18): E4151–E4158. doi: 10.1073/pnas.1719622115.
[4] CHEN J D, ZHENG C, MA J Q, et al. The chromosome-scale genome reveals the evolution and diversification after the recent tetraploidi- zation event in tea plant [J]. Hort Res, 2020, 7: 63. doi: 10.1038/s414 38-020-0288-2.
[5] WANG Y C, HAO X Y, LU Q H, et al. Transcriptional analysis and histochemistry reveal that hypersensitive cell death and H2O2have crucial roles in the resistance of tea plant [(L.) O. Kuntze] to anthracnose [J]. Hort Res, 2018, 5: 18. doi: 10.1038/s414 38-018-0025-2.
[6] LU Q H, WANG Y C, XIONG F, et al. Integrated transcriptomic and metabolomic analyses reveal the effects of callose deposition and multihormone signal transduction pathways on the tea plant-interaction [J]. Sci Rep, 2020, 10(1): 12858. doi: 10. 1038/s41598-020-69729-x..
[7] YANG H, LUO P G. Changes in photosynthesis could provide impor- tant insight into the interaction between wheat and fungal pathogens [J]. Int J Mol Sci, 2021, 22(16): 8865. doi: 10.3390/ijms 22168865.
[8] JIANG S L, YIN Q X, LI D X, et al. Integrated mRNA and small RNA sequencing for analyzing tea leaf spot pathogen, underconditions and the course of infection [J]. Phytopathology, 2021, 111(5): 882–885. doi: 10.1094/PHYTO-07-20- 0297-A.
[9] XU R F, YANG K, DING J, et al. Effect of green tea supplementation on blood pressure: A systematic review and meta-analysis of rando- mized controlled trials [J]. Medicine, 2020, 99(6): e19047. doi: 10. 1097/MD.0000000000019047.
[10] ASHRAFI-DEHKORDI E, ALEMZADEH A, TANAKA N, et al. Meta-analysis of transcriptomic responses to biotic and abiotic stress in tomato [J]. PeerJ, 2018, 6: e4631. doi: 10.7717/peerj.4631.
[11] PANAHI B, FRAHADIAN M, DUMS J T, et al. Integration of cross species RNA-seq meta-analysis and machine-learning models identifies the most important salt stress-responsive pathways in[J]. Front Genet, 2019, 10: 752. doi: 10.3389/fgene.2019.00752.
[12] WANG S S, LIU L, MI X Z, et al. Multi-omics analysis to visualize the dynamic roles of defense genes in the response of tea plants to gray blight [J]. Plant J, 2021, 106(3): 862–875. doi: 10.1111/tpj.15203.
[13] RAU A, MAROT G, JAFFRÉZIC F. Differential meta-analysis of RNA-seq data from multiple studies [J]. BMC Bioinform, 2014, 15: 91. doi: 10.1186/1471-2105-15-91.
[14] PANDIAN B A, SATHISHRAJ R, DJANAGUIRAMAN M, et al. Role of cytochrome P450 enzymes in plant stress response [J]. Antioxidants, 2020, 9(5): 454. doi: 10.3390/antiox9050454.
[15] FLORYSZAK-WIECZOREK J, ARASIMOWICZ-JELONEK M. The multifunctional face of plant carbonic anhydrase [J]. Plant Physiol Biochem, 2017, 112: 362–368. doi: 10.1016/j.plaphy.2017.01.007.
[16] BARROS J, SERK H, GRANLUND I, et al. The cell biology of lignification in higher plants [J]. Ann Bot, 2015, 115(7): 1053–1074. doi: 10.1093/aob/mcv046.
[17] WEI R F, LAI J D, PENG C B, et al. cDNA-AFLP reveals differential gene expression profiles of tea plant (cv. Maoxie) induced bysp.1 infection [J]. J Tea Sci, 2020, 40(1): 26–38. [魏日凤, 赖建东, 彭成彬, 等. 利用cDNA-AFLP技术分析炭疽菌危害诱导茶树的差异表达基因 [J]. 茶叶科学, 2020, 40(1): 26–38. doi: 10.3969/j.issn.1000-369X.2020.01.003.]
[18] LIU S Y, ZHANG Q Q, GUAN C F, et al. Transcription factor WRKY14 mediates resistance of tea plants [(L.) O. Kuntze] to blister blight [J]. Physiol Mol Plant Pathol, 2021, 155: 101667. doi: 10.1016/j.pmpp.2021.101667.
[19] BIAN Z Y, GAO H H, WANG C Y. NAC transcription factors as positive or negative regulators during ongoing battle between patho- gens and our food crops [J]. Int J Mol Sci, 2021, 22(1): 81. doi: 10. 3390/ijms22010081.
[20] IMANO S, FUSHIMI M, CAMAGNA M, et al. AP2/ERF transcription factor NbERF-IX-33 is involved in the regulation of phytoalexin production for the resistance ofto[J]. Front Plant Sci, 2022, 12: 821574. doi: 10.3389/ fpls.2021.821574.
[21] HUH S U. Functional analysis of hot pepper ethylene responsive factor 1A in plant defense [J]. Plant Signal Behav, 2022: 2027137. doi: 10. 1080/15592324.2022.2027137.
[22] ZHU Y T, HU X Q, WANG P, et al., an R2R3-type MYB transcription factor, positively regulates cotton resistance tothe lignin biosynthesis and jasmonic acid signaling pathway [J]. Int J Biol Macromol, 2022, 201: 580–591. doi: 10.1016/j. ijbiomac.2022.01.120.
[23] ZHU K K, WANG X L, LIU J Y, et al. The grapevine kinome: Annotation, classification and expression patterns in developmental processes and stress responses [J]. Hort Res, 2018, 5: 19. doi: 10.1038/ s41438-018-0027-0.
[24] FERREIRA-NETO J R C, DA COSTA B A N, DA SILVA M D, et al. The cowpea kinome: Genomic and transcriptomic analysis under biotic and abiotic stresses [J]. Front Plant Sci, 2021, 12: 667013. doi: 10. 3389/fpls.2021.667013.
[25] DING C Q, LEI L, YAO L N, et al. The involvements of calcium- dependent protein kinases and catechins in tea plant [(L.) O. Kuntze] cold responses [J]. Plant Physiol Biochem, 2019, 143: 190–202. doi: 10.1016/j.plaphy.2019.09.005.
[26] JEYARAJ A, WANG X W, WANG S S, et al. Identification of regulatory networks of microRNAs and their targets in response toin tea plant (L.) [J]. Front Plant Sci, 2019, 10: 1096. doi: 10.3389/fpls.2019.01096.
[27] JIN W B, WU F L. Identification and characterization of cucumber microRNAs in response toinfection [J]. Gene, 2015, 569(2): 225–232. doi: 10.1016/j.gene.2015.05.064.
[28] LI Y, LU Y G, SHI Y, et al. Multiple rice microRNAs are involved in immunity against the blast fungus[J]. Plant Physiol,2014, 164(2): 1077–1092. doi: 10.1104/pp.113.230052.
[29] YANG C C, WU P F, YAO X H, et al. Integrated transcriptome and metabolome analysis reveals key metabolites involved indefense against anthracnose [J]. Int J Mol Sci, 2022, 23(1): 536. doi: 10.3390/ijms23010536.
[30] JIA X L, WANG G L, XIONG F, et al.assembly, trans- criptome characterization, lignin accumulation and anatomic character- ristics: Novel insights into lignin biosynthesis during celery leaf development [J]. Sci Rep, 2015, 5: 8259. doi: 10.1038/srep08259.
[31] LIU X T, CAO X Q, SHI S C, et al. Comparative RNA-Seq analysis reveals a critical role for brassinosteroids in rose () petal defense againstinfection [J]. BMC Genet, 2018, 19(1): 62. doi: 10.1186/s12863-018-0668-x.
[32] YANG R, JIANG S L, LI D X, et al. Integrated mRNA and small RNA sequencing for analyzing leaf spot pathogenand its host, tea (), during infection [J]. Mol Plant Microbe Interact, 2021, 34(1): 127–130. doi: 10.1094/MPMI-07-20- 0207-A.
[33] ZHANG Y, ZHANG A H, LI X M, et al. The role of chloroplast gene expression in plant responses to environmental stress [J]. Int J Mol Sci, 2020, 21(17): 6082. doi: 10.3390/ijms21176082.
[34] KANGASJÄRVI S, TIKKANEN M, DURIAN G, et al. Photosynthetic light reactions: An adjustable hub in basic production and plant immunity signaling [J]. Plant Physiol Biochem, 2014, 81: 128–134. doi: 10.1016/j.plaphy.2013.12.004.
[35] SHASMITA, SAMAL P, MOHAPATRA P K, et al. Improved photo- system II and defense enzymes activity in rice () by biopriming againstpv.[J]. Funct Plant Biol, 2021, 48(3): 298–311. doi: 10.1071/FP20221.
[36] BWALYA J, ALAZEM M, KIM K H. Photosynthesis-related genes induce resistance against soybean mosaic virus: Evidence for involve- ment of the RNA silencing pathway [J]. Mol Plant Pathol, 2022, 23(4): 543–560. doi: 10.1111/mpp.13177.
[37]LIU B G. Transcription regulation of lignin biosynthesis and interaction of monolignol biosynthesis enzymes in[D]. Harbin: Northeast Forestry University, 2020. [刘宝光. 毛果杨木质素生物合成的转录调控及单体生物合成酶互作研究 [D]. 哈尔滨: 东北林业大学, 2020.]
[38] LIN CY, SUN Y, SONG J N, et al. Enzyme complexes of Ptr4CL and PtrHCT modulate co-enzyme a ligation of hydroxycinnamic acids for monolignol biosynthesis in[J]. Front Plant Sci, 2021, 12: 727932. doi: 10.3389/fpls.2021.727932.
[39] LI X, ZHANG Y Y, ZHAO S J, et al. Omics analyses indicate the routes of lignin related metabolites regulated by trypsin during storage of pitaya () [J]. Genomics, 2021, 113(6): 3681– 3695. doi: 10.1016/j.ygeno.2021.08.005.
[40] VÖGELI B, ENGILBERGE S, GIRARD E, et al. Archaeal aceto- acetyl-CoAthiolase/HMG-CoA synthase complex channels the inter- mediate via a fused CoA-binding site [J]. Proc Natl Acad Sci USA, 2018, 115(13): 3380–3385. doi: 10.1073/pnas.1718649115.
[41] ZHENG T, GUAN L B, YU K, et al. Expressional diversity of grape- vine 3-hydroxy-3-methylglutaryl-CoAreductase () in different grapes genotypes [J]. BMC Plant Biol, 2021, 21(1): 279. doi: 10.1186/s 12870-021-03073-8.
Common Pattern in Response to Pathogenic Fungal Stress of Tea Plants Based on Meta-analysis
LIAN Lingli1a, CHEN Qiang1b, ZHOU Ying1a, FU Jing1a, LI Wanying1a, WEI Rifeng1b, LIU Wei2*
(1a. College of Life Sciences; 1b. College of Horticulture, Fujian Agriculture and Forestry University,Fuzhou 350002, China; 2. Ningde Normal University, Ningde 352100, Fujian, China)
To explore the common response mode and disease resistance mechanism of tea plants () to pathogenic stress, bioinformatics methods were used to extract, integrate and function enrich of multiple sets of RNA-seq data, and the main regulatory molecules and protein interaction modules were analyzed by combining various tools and database resources. This results showed that the expression of cytochrome P450 family members in tea plant was significantly up-regulated under the fungal pathogen stress. The metabolic processes of steroid and hormone, and phenylpropanoid synthesis pathway were activated, and the biological processes, such as mitotic cell cycle regulation, DNA methylation and photosynthesis pathway were inhibited. The major regulatory molecules, such asandtranscription factors, theandfamily of kinases were mainly up-regulated. The differentially expressed protein interaction modules showed that the modules involved in mitotic cycle regulation, microtubule motion-based, starch and sucrose metabolism, cell wall polysaccharide synthesis, photosynthesis, flavonoid metabolism were down-regulated, while lignin synthesis and terpenoid biosynthesis were up-regulated. There may be interactions between modules. The key genes in lignin and terpenoid synthesis pathways activated by pathogen stress included ferulic acid-5-hydroxylase gene (), peroxidase gene () and terpenoid synthase gene. Cytochromegene might play a key role in fungus stress of tea plants. Enhancing the synthesis of lignin and terpenoids, and weakening photosynthesis might be the core modes of tea plants responding to fungus stress.
Tea plant; Fungal pathogen; Pathogenic fungi stress; Meta-analysis
10.11926/jtsb.4628
2022-02-24
2022-03-27
福建农林大学科技创新专项基金项目(KFA20047A, KFA20143A);福建农林大学大学生创新项目(202110389107)资助
This work was supported by the Project for Science and Technology Innovation in Fujian Agricultre and Forestry University (Grant No. KFA20047A, KFA20143A), and the Project for Undergraduate Innovation in Fujian Agricultre and Forestry University (Grant No. 202110389107).
连玲丽(1979年生),女,博士,副教授,研究方向为逆境生物信息学。E-mail: lianll2002@163.com
E-mail: liuwei0593@126.com
猜你喜欢
小哥白尼(军事科学)(2019年9期)2019-12-21
电影(2019年3期)2019-04-04
乡村地理(2018年2期)2018-09-19
特别健康(2018年3期)2018-07-04
少儿科学周刊·少年版(2017年3期)2017-06-29
广东农业科学(2016年2期)2016-07-13
衡阳师范学院学报(2016年3期)2016-07-10
湖南农业(2016年3期)2016-06-05
学生天地(2016年14期)2016-05-17
音乐天地(音乐创作版)(2016年11期)2016-02-05
