
面向溢油污染治理的SiO2 气凝胶疏水改性的研究进展
2023-03-13 08:16张添华肖龙恒
工程科学学报 2023年6期
韩 松,张添华,肖龙恒,郭 敏,张 梅✉
1) 北京科技大学冶金与生态工程学院,北京 100083 2) 中冶建筑研究总院有限公司,北京 100088
化合物缩写目录
本文所用到的化合物缩写名称如下表所示.

文中所用到的化合物缩写名称表List of abbreviated names of compounds used in this paper
石油开采、加工和运输过程中的石油泄漏污染严重危害了人类和生态系统的健康[1].例如,墨西哥湾的石油泄漏,是历史上最大的原油泄漏事故,约有490 万桶石油泄漏到海洋中,对环境和经济造成严重破坏[2].油污中很多有毒的有机化合物在水中难以降解,长期累积在海洋生物中,最终通过食物链危害人类健康[3].因此,为了减少或避免石油污染对生态环境的长期污染,迫切需要开发一种用于有效清除溢油污染的吸油材料[4].目前溢油污染的处理方法中,燃烧法会造成环境污染,消耗能源[5];分散剂、固化剂对水生生物有害、成本高[6];撇油器、围油栏耗时而且成本非常高,处理效果不佳;微生物法降解速度缓慢[7].吸附法不仅成本低、处理高效、易于回收和适应性好,而且对环境友好,因而成为溢油污染处理的研究热点[8-10].传统的吸油材料中,天然有机吸附材料吸油能力低、亲水;无机吸附材料回收困难,吸油效率低、价格昂贵;合成有机吸附材料虽然吸油能力突出,但不可生物降解[11].因此,亟需开发一种成本低、疏水性好、吸附能力强、可循环使用的新型吸油材料.
SiO2气凝胶是由纳米粒子相互堆积组成的一种高孔隙率(~99.8%)、低密度(~0.003 g·cm-3)、高比表面积(1200 m2·g-1)、低热导率(~0.005 W·(m·K)-1)的三维网状多孔材料[12-13].这些特性使其在航空航天(切伦科夫探测器辐射器[14])、隔热[15]、吸附(重金属离子[16]、油和有机液体等[17])、催化剂载体[18]等领域有广阔的应用前景.然而,传统溶胶-凝胶法制备的SA 表面亲水,不适用于从水和油混合物中吸附油.
经过疏水改性后的SA 除了具有密度低、比表面积大、孔隙率高等特点外,还具有独特的憎水/亲油性,非常适用于吸油材料[19],近年来成为吸油材料方面研究的热点.HSA 的制备方法主要有表面后处理法、共前驱体法以及气相氧化法,其中常用的是表面后处理法和共前驱体法.
本文主要围绕表面后处理法和共前驱体法,介绍了两种方法结合超临界干燥和常压干燥制备HSA 的研究进展,对比总结了它们的优缺点.此外,本文还介绍了目前HSA 作为吸油材料的吸油机理、吸油性能、混合油的吸附及吸油循环使用性能(再生性).
1 SiO2 气凝胶疏水改性
大多数传统的SA 是以硅醇盐(Si(OR)4)(如正硅酸甲酯(TMOS,Si(OCH3)4)、正硅酸四乙酯(TEOS,Si(OCH2CH3)4))为硅源制备的,表面大量羟基(—OH)的存在,使其极易因吸附水分而发生开裂、坍塌,结构稳定性变差[20],不利于在吸油领域上的应用.因此,为消除SA 表面的羟基对其结构和吸油性能的影响,需要对其进行表面疏水改性处理.目前SA 常用的改性方法是表面后处理法和共前驱体法.此外,也有少量HSA 为气相氧化法制备.
1.1 SiO2 气凝胶的制备
SA 是研究最早的一类气凝胶,也是目前研究最广泛和最常用的气凝胶类型.1931 年,Kistler[21]首次采用溶胶-凝胶工艺,以Na2SiO3为硅源,结合超临界干燥制备了SA.目前,SA 一般采用溶胶-凝胶工艺制备湿凝胶,如图1 所示.具体工艺可分为两步:(1)溶胶-凝胶:在催化剂作用下,硅源水解,水解产物发生羟基缩聚反应形成湿凝胶,再经过老化过程,来强化湿凝胶的网络结构.在溶胶-凝胶过程中,首先形成的初级粒子聚集形成次级粒子,最后形成相互缠绕的珍珠项链结构[22].(2)干燥:通过干燥工艺去除湿凝胶中的溶剂,得到SA[23].其中,超临界干燥和常压干燥是常用的干燥工艺[20].超临界干燥可以生产较大块SA,但是其设备昂贵,工艺复杂,危险性大[24].而常压干燥设备便宜,工艺简单,是目前SA 干燥工艺的主要研究方向.

图1 SA 的制备流程和反应机理Fig. 1 Preparation process and reaction mechanism of SA
1.2 SiO2 气凝胶疏水改性机理和方法
SA 疏水改性的主要机理为:疏水性基团与SA 表面的羟基发生取代反应,疏水基团取代亲水性羟基(—OH),再经过老化、溶剂置换以及干燥工艺制备HSA[25].目前,SA 疏水改性的主要方法有表面后处理法和共前驱体法[26-27],此外还有气相氧化法[28].
(1)表面后处理法.
表面后处理法(又称表面衍生法)是制备HSA的常用方法,也是最早用于气凝胶疏水改性的方法.表面后处理法的改性对象是溶胶-凝胶过程得到的湿凝胶,经过溶剂置换后,将其浸泡在疏水改性液中,与湿凝胶表面的羟基反应,完成疏水改性.图2 为TMCS 疏水改性SA 的过程及反应机理.表面后处理法常用三甲基氯硅烷(TMCS)、甲基三甲氧基硅烷(MTMS)和六甲基二硅氮烷(HMDZ)等[29-30]硅烷化试剂对SA 疏水改性.

图2 TMCS 表面后处理法制备HSA 的改性过程及机理[34]Fig. 2 Modification process and mechanism of HSA prepared by TMCS surface post-treatment[34]
(2)共前驱体法.
共前驱体法(又称原位法、共聚法),是指将含有疏水基团的前驱体与硅醇盐作为共前驱体,硅醇盐水解缩合后改性剂也水解缩合,疏水基团嫁接在SA 表面上,在溶胶-凝胶过程中完成表面改性,进而实现SA 的疏水改性[31].以三甲基乙氧基硅烷(TMES)/正硅酸甲酯(TMOS)为共前驱体疏水改性SA 的机理为[32]:
水解反应:
缩合反应:
TMOS 先水解缩合使得溶胶表面附着大量的羟基,然后TMES 开始水解缩合.式(1)中,TMES水解生成(CH3)3Si—OH.由式(2)~(3)可知,TMES与溶胶、水解产物与溶胶表面的羟基发生缩合反应生成疏水的—O—Si(CH3)3,从而实现SA 的原位改性.
(3)气相氧化法.
表面后处理法和共前驱体法都是在液相中完成改性,而气相氧化法是在高温甲醇蒸汽气氛中完成改性,即甲醇蒸汽进入湿凝胶孔隙中发生甲氧基化反应,使得硅羟基(Si—OH)转变为Si—O—CH3基,完成疏水改性[33].Anderson 等[28]以TMOS、MTMS、乙基三甲氧基硅烷(ETMS)和丙基三甲氧基硅烷(PTMS)为共前驱体的湿凝胶在高温甲醇蒸汽中完成甲氧基化反应,制备了HSA,其水接触角为155°.
气相氧化法需要在超临界条件下进行,设备复杂,成本高,这很大程度上限制了在疏水改性SA 上的应用,因此,本文不作详细介绍.现阶段,共前驱体法和表面后处理法还是SA 最主要的疏水改性方法.根据其含的有机官能团的数量,可分为单官能硅烷化剂、双官能硅烷化剂和三官能硅烷化剂,如表1 所示.

表1 三种官能硅烷剂的分类[35-36]Table 1 Classification of monofunctional,difunctional,and trifunctional silylating agents[35-36]
2 表面后处理法制备疏水SiO2 气凝胶
表面后处理法是获得HSA 最常用的方法.采用表面后处理法疏水改性的效果往往会受到硅源、催化剂、置换溶剂、改性剂及其含量、干燥等因素的影响[33].表面后处理法常用的硅源有TMOS、TEOS.催化剂包括碱性催化剂(氢氧化钠、氨水、氟化铵/氨水等)和酸性催化剂(盐酸、氢氟酸、草酸等).常用的溶剂为乙醇、丙酮、正己烷等.常用的疏水改性剂如表1 所示.表面后处理法常结合超临界干燥和常压干燥制备HSA.
2.1 表面后处理法—超临界干燥制备疏水SiO2气凝胶
超临界干燥是研究气凝胶干燥最早的工艺[37].在临界温度和临界压力下,气-液界面消失,溶剂基本上不存在表面张力,在干燥过程时SA 不会发生收缩和坍塌,因而可以获得极高孔隙率、超低密度的高品质SA[38].超临界干燥介质有二氧化碳、甲醇、乙醇,其中CO2是最常用的干燥介质[39].超临界干燥是表面后处理法制备HSA 的干燥手段之一.
卢斌等[40]以TEOS 和异丙醇为原料,HMDZ和TMCS 为表面改性剂,超临界干燥制备了HSA.其反应机理为:
由式(4)可知,加入改性剂TMCS 后,—Si(CH3)3基团接枝于二氧化硅团簇表面从而替代了表面的Si—OH,随着正向反应程度的增加,二氧化硅团簇表面的—CH3数量增加,疏水性增大,其水接触角为157°.式(5)~(6)为HMDZ 改性反应机理,其反应分两步进行,用HMDZ 改性后的样品水接触角为120°.
Wang 等[41]采用一种新的硅源,以聚二乙氧基硅氧烷(PDEOS)为硅源,TMCS 和二甲基二甲氧基硅烷(DMMOS)为硅烷化剂,结合超临界干燥得到HSA.与TMOS 和TEOS 相比,PDEOS 制备的HSA 的聚合度更高.根据式(7)~(8)可知,与TMCS相比,DMMOS 表面改性效果更佳.
TMCS 改性:
DMMOS 改性:
超临界干燥是表面后处理法减少或避免HSA干燥时结构破裂的有效方法.超临界干燥工艺仍然是获得高品质、高性能大块SA 的最佳干燥工艺.
2.2 表面后处理法—常压干燥制备疏水SiO2 气凝胶
超临界干燥设备条件苛刻,危险性大,成本高.而常压干燥设备简单,成本低,生产周期短.因此,常压干燥是HSA 实现低成本、连续化和规模化生产的主要研究方向[42].目前已经在硅源的种类、改性剂种类及其浓度、置换溶剂、干燥过程等方面制备HSA 进行了大量的研究.
刘洋等[43]以TEOS 为硅源,TMCS/乙醇为表面后处理改性液,常压干燥制备了HSA.根据FTIR表明,改性后凝胶骨架表面的—OH 被Si(CH)3—O—所取代,从而得到HSA.其水接触角高达158°,比表面积为911.67 m2·g-1.
当前,表面后处理法常采用TMOS、TEOS 为硅源,但是其成本较高,很大程度上限制了工业化生产,表2 为列出了目前市面上硅源和改性剂的销售价格,从图中可以看出,纯试剂TMOS、TEOS价格昂贵,而高岭土等原料价格相对低廉,可作为低成本的替代硅源.为了降低生产成本,He 等[44]以水玻璃为硅源,以15%的TMCS 和85%的正己烷(体积分数)为疏水改性液,结合常压干燥制备了HSA,其水接触角达到165°,比表面积为817 m2·g-1,密度低至0.095 g·cm-3.此外,我国高岭土资源丰富,其中二氧化硅含量高,价格低,是制备HSA 的理想原料,Hu 等[45]以高岭土为主要原料,体积比为1.5 的TMCS 和正己烷溶液为改性液,常压干燥制备得到HSA.其比表面积为465.03 m2·g-1,水接触角为152°.

表2 硅源及改性剂的成本Table 2 Cost of silicon source and silylating agents
卢斌等[46]以硅溶胶为硅源,四种酸(盐酸、硝酸、乙酸、草酸)作为催化剂,以TMCS/正己烷为改性液,采用常压干燥制备了HSA.研究发现,以草酸作为催化剂时,其较大的分子构成阻碍了溶胶颗粒间的堆积,防止过大粒子的形成,从而使得HSA 结构均匀,具有较好的结构和性能,其表观密度为0.157 g·cm-3、比表面积为542.1 m2·g-1、水接触角为153.8°.
常压干燥前,需要用表面张力低的溶剂多次替换掉凝胶孔洞中的水,降低SA 在常压干燥中凝胶受到的毛细管力.正己烷(C6H14,18.40×10-7N·m-1的表面张力很低[47],适合用作置换溶剂.毕海江等[48]以TEOS 为前驱体,酸碱两步溶胶-凝胶法制备了湿凝胶,老化后的湿凝胶在无水乙醇中置换1 次,正己烷置换2 次,以TMCS 为改性剂,常压干燥后制备了HSA.其水接触角达到158°,比表面积高达1067 m2·g-1.此外,Rao 等[47]研究了不同置换溶剂(正己烷、环己烷、庚烷、苯、甲苯、和二甲苯)对HSA 疏水性和物理性质的影响.研究发现,庚烷置换得到的HSA 孔隙率最高(97%)、比表面积最大(750 m2·g-1)、低密度最低(~0.060 g·cm-3),水接触角达到160°,整体性能最好.
然而,多次溶剂置换和表面改性过程是单独进行,制备周期长,溶剂消耗量大.有研究[49]采用一步法(溶剂交换的同时完成表面改性),该方法多采用TMCS 为疏水改性剂[50].Lee 等[51]以水玻璃为原料,将制备的湿凝胶浸入异丙醇(IPA)/TMCS/正己烷组成的溶液中,同时完成溶剂置换和表面改性处理,得到了块状HSA.其反应机理如图3 所示.该气凝胶孔隙率为94%,比表面积达到630 m2·g-1.Bhagat 等[52]以水玻璃为硅源,在硝酸/HMDZ/正己烷溶液中同时完成溶剂交换和表面改性,制备得到HSA 粉末.

图3 HSA 在IPA/TMCS/正己烷中的反应机理[51]Fig. 3 Reaction mechanism of HSA in IPA/TMCS/n-Hexane solution[51]
一步法缩短了制备周期,溶剂消耗量减少.但是,其改性过程不容易控制,疏水基团分布不均匀,低表面张力溶剂替换原母液时,产生的表面张力梯度会破坏气凝胶的骨架结构,在常压干燥时易造成SA 开裂.
不同官能硅烷改性剂的改性效果各有不同.Mahadik 等[35]以单官能TMCS、双官能DMDCS 和三官能MTMS 为硅烷化改性剂,研究了不同官能团硅烷试剂对SA 理化性质的影响.研究结果表明,TMCS、DMDCS 和MTMS 改性的SA 样品水接触角分别为(161±2)°、(152±3)°和(147±3)°.根据FTIR和反应机理,与DMDCS 和MTMS 硅烷化相比,TMCS的甲基取代了凝胶表面更多的羟基,表面硅烷化更有效.与双官能DMDCS 和三官能MTMS 相比,单官能TMCS 改性的SA 表现出更高的热稳定性、更高比表面积、更高的水接触角以及更高的孔隙率.
TMCS 和HMDZ 是常用的单官能表面改性剂,但是TMCS 改性的SA 综合性能更加优异[48].Shewale 等[53]研究发现,TMCS 改性效果更好.HMDZ的改性反应机理为:
式(9)中反应生成的三甲氨基硅烷可以与Si—OH继续反应,如式(10),降低了HMDZ 与硅羟基的反应速率.FTIR 分析得出,与HMDZ 相比,TMCS 改性的SA 在840 cm-1处Si—C 伸缩振动引起的吸收峰和在2900 cm-1处C—H 弯曲振动引起的吸收峰较强,说明TMCS 改性后表面接上了更多的甲基,改性效果更佳,其密度低至0.04 g·cm-3,水接触角为149°,而HMDZ 改性的SA 的密度为0.1 g·cm-3,水接触角为143°.即使改性剂的疏水基团相同,但是硅烷化剂的性质不同,得到的SA 的物理性质也不同.此外,Mahadik 等[54]研究了不同浓度的TMCS和HMDZ 对SA 改性效果的影响,FTIR 分析发现,随着硅烷化试剂浓度从3%增加到12%,非极性—CH3基团取代了更多极性—OH 基团,SA 的疏水性增加.TMCS、HMDZ 改性后的HSA 水接触角分别从128°、123°增加到156°、155°.
有研究表明,硅烷化剂的组合(CSMA,Combinations of surface modification agents)不仅能够减少SA干燥时发生的体积收缩,而且能够增强凝胶的表面改性效果.Rao 等[55]以HMDSO/TMCS、HMDSO/HMDZ 和HMDZ/TMCS 为混合硅烷化剂改性SA,其表面改性反应如图4 示.单独采用TMCS 改性时,反应产生的HCl 使得凝胶结构未能均匀排列,而HMDSO 和HMDZ 改性都是两步过程(两个—Si(CH3)3)基团双重离解),过程缓慢,而且HMDSO 产物之一的水会抑制改性的进行.结果表明,相对于单一硅烷化剂改性,CSMA 改性后的气凝胶结构更均匀、密度更低、孔隙率更高、孔隙体积更大以及更稳定的低表面能.由图4 可知,TMCS/HMDSO 组合改性时,HCl 的生成使得HMDSO 活性增加,并且可以与HMDSO 反应生成更多的TMCS.因此,TMCS/HMDSO 组成的CSMA 表面改性能力更佳,改性后的气凝胶水接触角高达154°,密度低至0.042 g·cm-3,并且在425 ℃下仍能保持疏水性.
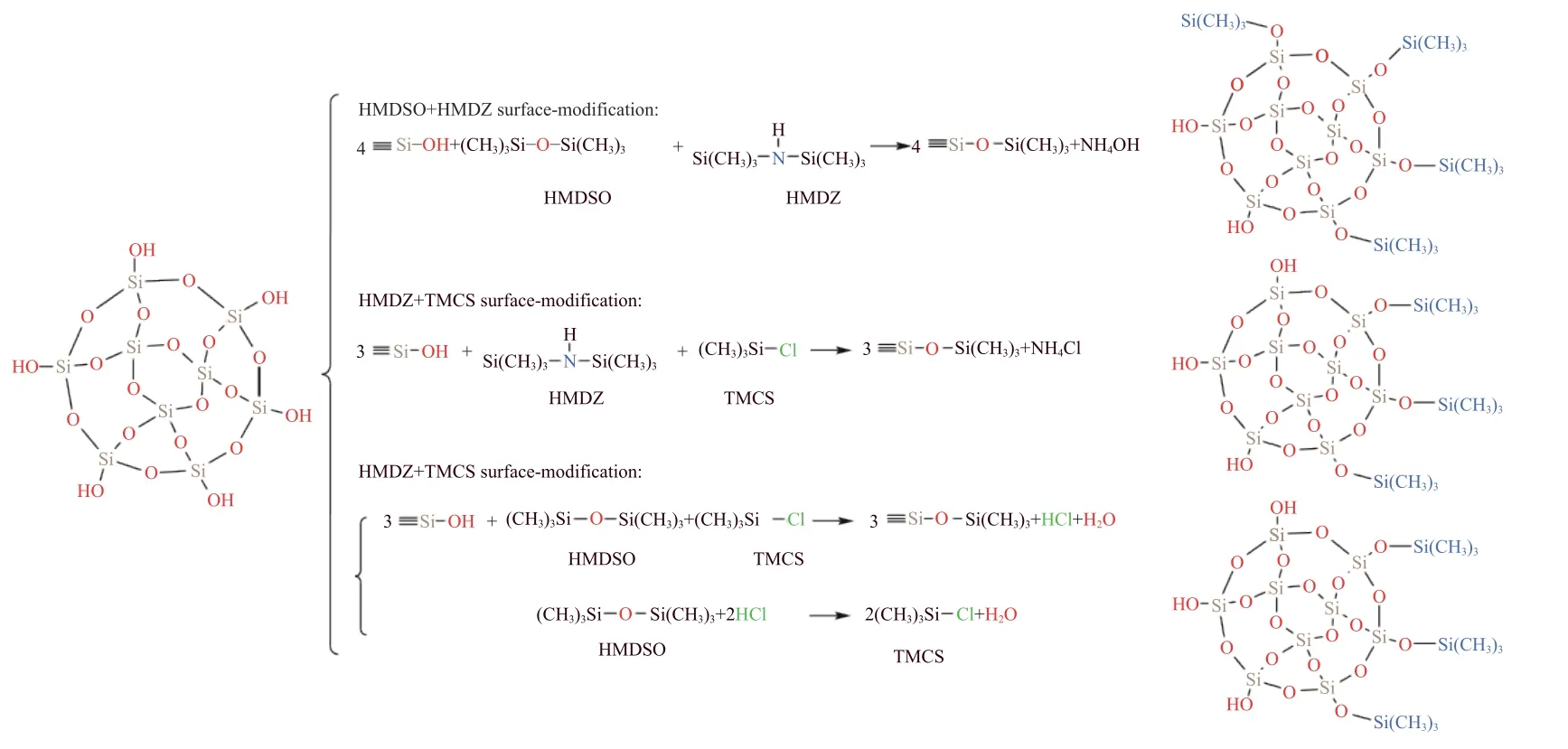
图4 CSMA(HMDSO/TMCS、HMDSO/HMDZ 和HMDZ/TMCS)改性SA 的反应机理[55]Fig. 4 Reaction mechanism of CSMA (HMDSO/TMCS,HMDSO/HMDZ,and HMDZ/TMCS) modified SA[55]
研究发现,相比于单次表面疏水改性,多次表面改性(MSM,Multiple surface modification)的效果更佳.Wei 等[56]以TMCS 为改性剂,经过4 次表面改性后,得到的HSA 孔隙率高达96.8%,比表面积为777 m2·g-1,水接触角达到143°.此外,常压干燥后样品的体积收缩率仅为1%.
常压干燥的SA 容易发生开裂,在凝胶形成前添加干燥控制化学添加剂(DCCA,Drying control chemical additives)后,可减少或避免气凝胶在干燥中体积收缩和开裂[57].DCCA 抑制了凝胶颗粒的生长,可以获得更均匀的粒径和孔径分布[58].常用的DCCA 有甲酰胺、乙酰胺、N,N-二甲基甲酰胺(DMF)、N,N-二甲基乙酰胺等[59],其中DMF 使用较多.Liu 等[58]以TEOS 为硅源,DMF 为DCCA,IPA/TMCS/正己烷为改性液,研究了DMF/TEOS 摩尔比对HSA 物理性质的影响.DMF 的加入降低了气凝胶的表面张力,获得更均匀粒径和更小的凝胶颗粒.当DMF/TEOS 摩尔比0.8 时,HSA 收缩率最低(6.32%),水接触角高达178°,吸水率仅为5.1%.
目前,为了减少或避免HSA 在常压干燥时发生收缩和坍塌的主要方法有:(1)增强凝胶骨架机械强度,如延长老化时间来增强SA 骨架[60-61]、添加DCCA[57,62]等;(2)选择表面张力低的溶剂置换,减少毛细管力的影响,如采用正己烷、丙酮、环己烷、庚烷等溶剂[47].
随着常压干燥技术的发展,常压干燥可以作为代替超临界干燥的有效方法.Torres 等[29]以7 种硅烷化剂,分别在常压干燥和超临界干燥下制备了HSA,研究各种改性剂和两种干燥条件对SA 性能的影响.结果表明,常压干燥过程中气凝胶结构发生重排,使其孔隙结构更佳发达、强度更高.常压干燥的HSA 接触角大于139°,尽管超临界干燥的HSA 接触角更大(>169°),但在常压干燥下,TMMS、TMES、HMDZ 这3 种硅烷试剂制备气凝胶的综合改性效果都很好,可以满足对HSA 性能的需求.
目前,采用表面后处理法制备HSA 的研究比较多,且常结合常压干燥工艺使用.该方法是在形成湿凝胶后进行改性,改性不影响原有的孔隙结构,方法简单,容易控制,但是要经过多次溶剂置换,工艺繁琐,成本高,可能存在HSA 内部改性不彻底的问题.
3 共前驱体法制备疏水SiO2 气凝胶
共前驱体法是采用含疏水基团的硅烷化剂(如MTMS、MTES 等),来部分或全部替代传统前驱体(TMOS、TEOS 等)制备HSA 的方法[33].与表面后处理法相比,共前驱体法的疏水改性和湿凝胶的形成过程同时进行,湿凝胶形成后无需表面改性处理,制备流程短,节约了成本.此外,疏水基团的引入还会降低凝胶内部的刚性连接,增加SA的柔性.共前驱体方法由Schwertfeger 等[63]首创,以TMOS/MeTMOS 为共前驱体,结合超临界干燥制备了HSA.共前驱体法常用的硅源有TMOS、TEOS、MTMS 和MTES 等[64].
3.1 共前驱体法—超临界干燥制备疏水SiO2气凝胶
目前,共前驱体法常以TMOS 或TEOS 为硅源,MTMS 为共前驱体制备HSA[27].吴国友等[65]以TEOS/MTMS 为共前驱体改性剂,结合超临界干燥,得到块状HSA.相比于未改性前,改性后的SA密度较低(0.1 g·cm-3)、比表面积较高(1070 m2·g-1)、孔隙率较大(95.5%),接触角从110°增加到140°,疏水耐温性从250 ℃升高到500 ℃,热稳定性更好.
共前驱体的种类及其加入比例对HSA 的性能有很大影响.Rao 等[32]研究了不同摩尔比的TMES/TMOS 作为共前驱体,在超临界干燥的条件下制备HSA.研究发现,随着TMES/TMOS 摩尔比增大,TMES 中更多的—Si(CH3)3基团附着到气凝胶表面,气凝胶的疏水性增大.当TMES/TMOS 摩尔比为2.5 时,HSA 的水接触角为130°.Yu 等[39]以MTES/TEOS 为共前驱体,采用酸碱两步催化法,结合超临界干燥,制备了HSA.当MTES/TEOS 摩尔比增加到0.8 时,得到超疏水SiO2气凝胶(接触角为152°),比表面积最大为996.35 m2·g-1.
改性剂本身的性质以及共前驱体之间的反应程度同样会极大地影响改性效果.Rao 等[30]以六种RnSiX4-n型共前驱体改性剂(MTMS、MTES、DMCS、TMES、ETES 和PTES),结合超临界干燥得到HSA.研究发现,DMCS 基气凝胶的高度酸性使HSA 更容易收缩,其体积收缩率最高,达到44%,并且有裂纹.如表1 所示,有机官能团(R)的类型和R/Si 的比(烷基R 和Si 原子的个数比)是影响气凝胶结构的重要因素.TMES 共前驱体存在更多的有机基团(R/Si 比为3),从而增加了气凝胶的弹性,使得TMES 共前驱体制备的气凝胶收缩率最小(<2%).所有HSA 的接触角在120°~136°之间,其中TMES 共前驱体的水接触角最高(136°).
MTMS、MTES 等共前驱体制备的HSA 仍存在力学性能较差的问题,为提高其力学性能,采用功能化的有机共前驱体是一个有效方法.Parale 等[66]以TEOS 为主硅源,3-(三甲氧基硅基丙基)甲基丙烯酸酯(TMSPM)为共前驱体,结合超临界干燥,制备了疏水、热稳定性好、机械性能好、导热系数低的HSA.当添加质量分数为30%的TMSPM 时,HSA 疏水性最好(水接触角为140°),低导热系数最低(0.038 W·(m·K)-1),硬度增加到0.15 GPa.相比于未改性前,改性后的气凝胶热稳定性更好,在400 ℃下,该气凝胶仍然保持疏水特性.
共前驱体法引入的疏水基团有限,过量的疏水基团将会破坏气凝胶的结构.Ren 等[67]开发了一种新的合成方法,以聚乙氧基二硅氧烷为硅源,MTMS 为疏水改性剂,MTMS/乙醇溶液作为超临界干燥介质,在干燥过程中完成改性,避免了疏水基团对气凝胶结构的影响.得到的HSA 水接触角为146°,体积收缩率最低为13%,比表面积571.1m2·g-1.
共前驱体法中前驱体试剂的加入量不容易控制,引入过量的疏水基团,会影响SA 的结构.因此,结合超临界干燥,可以减少或避免SA 在干燥时发生结构坍塌和收缩,制备结构完整的HSA.
3.2 共前驱体法—常压干燥制备疏水SiO2 气凝胶
共前驱体法结合常压干燥也是制备HSA 重要手段之一.陈宇卓等[68]以水玻璃为主硅源,MTMS为共前驱体,乙醇、MTMS、水玻璃的摩尔比为12∶3∶1 时,常压干燥得到性能良好的超疏水SiO2气凝胶,其密度为0.097 g·cm-3,比表面积高达813.28 m2·g-1,水接触角高达150.3°.
Sharad 等[69]采用溶胶-凝胶法,以MTMS 为共前驱体疏水改性剂,结合常压干燥制备了MTMS基HSA.改性后,—CH3基团附着在凝胶骨架上,干燥时,甲基之间相互排斥,克服毛细管压力,发生“回弹效应”,从而使得高度多孔的HSA 结构不坍塌.其密度低至0.062 g·cm-3、比表面积为520 m2·g-1、水接触角高达152°,表现出超疏水性.
干燥控制化学添加剂(DCCA)仍然是共前驱体法减少或避免SA 在常压干燥中发生开裂和体积收缩的有效方法.李贵安等[70]以TEOS 为硅源,TMCS 为共前驱体,DMF 为DCCA,结合常压干燥制备了块状HSA.其比表面积可达979 m2·g-1,粒径小,疏水性好,DCCA 的加入可以提高气凝胶的整体性能.Cai 等[71]以MTMS 为前驱体,DMF 为DCCA,研究了DMF 对SA 物理性能的影响.如图5所示,DMF 的醛基通过形成的氢键吸附在二氧化硅簇上,从而抑制了MTMS 的水解和缩合反应,延长了凝胶时间.DMF 改性的气凝胶孔径分布更窄、更均匀.当DMF/MTMS 摩尔比为0.6 时,SA具有低密度、大比表面积、高孔隙率、良好的疏水性、均匀的孔径等优异特性.

图5 DMF 作为DCCA 的反应机理(R 为疏水基团)[71]Fig. 5 Reaction mechanism of DMF as DCCA (R-hydrophobic group)[71]
表面后处理法和共前驱体法是SA 疏水改性的主要方法,其制备流程及机理如图6 所示,两种方法各有特点.共前驱体法主要结合超临界干燥工艺制备HSA,采用常压干燥制备的研究较少.表面后处理法则常采用常压干燥.表3 为两种方法的对比.表面后处理法的对象是已形成的湿凝胶,改性不影响已形成的凝胶孔隙结构,HSA 的孔径和粒径比较均匀,但是可能存在内部改性不彻底的问题[72],另外多次溶剂置换需要消耗大量的溶剂,工艺繁琐,成本高.共前驱体法处理是在溶胶-凝胶过程中初生成的原生粒子和生长的聚集体上完成疏水改性,凝胶的形成和疏水改性过程同时进行,缩短了流程,所需的改性剂量很少.相比于表面后处理法,共前驱体法是表面和本体改性,改性得到的HSA 比表面积和孔体积较大[70],疏水性更好[30,73],更有利于在吸附方面的应用.但共前驱体法获得的HSA 孔径不均匀[31],引入的疏水基团有限,过量的疏水基团将影响HSA 湿凝胶的形成,制得的HSA 强度低[74].两种方法各有优缺点,为了最大化二者的优点,有研究同时采用硅烷化剂(如HMDZ)作为共前驱体法和表面后处理法的改性剂[75],得到性能更好的HSA,但是相关报道较少.

表3 表面后处理法和共前驱体法对比Table 3 Comparison between the surface posttreatment method and coprecursor method
4 疏水SiO2 气凝胶机械性能的增强
珍珠项链结构的SA 颗粒间颈部连接很弱[76],因此,无论是表面后处理法还是共前驱体法制备的HSA 都存在机械性能差的问题,不利于HSA 从油水混合物中吸附油.为了提高其机械性能,常用的解决办法有前驱体增强(结构性增强)、物理增强(SA 复合材料增强[77])以及化学增强(引入有机聚合物[78-79])等.
前驱体增强主要是采用含有机基团的前驱体(如MTMS、MTES),减小在干燥过程中毛细管力对SA 结构的破坏,提高气凝胶的力学性能.而且前驱体中疏水基团的引入使得HSA 在外力作用下,疏水有机基团间相互排斥,降低了凝胶内部的刚性连接,从而增强了HSA 骨架的机械强度.Rao等[4]以MTMS 为前驱体,利用溶胶-凝胶法,结合超临界干燥制备了柔性和高弹性的HSA.其水接触角为162°,杨氏模量低至1.094×104N·m-2,呈现出高度可压缩性和弹性.
物理增强主要是在HSA 中添加增强相,使其充当骨架支撑气凝胶所承受的机械负荷.纤维作为增强相是提高SA 机械性能的有效方法[80-81],用于增强SA 强度的纤维有玻璃纤维[82]、陶瓷纤维[83]、碳纳米纤维[84]以及聚合物纤维[85]等.Li 等[86]以玻璃纤维薄膜为增强相,制备了弹性和柔韧性的HSA,使其能够承受大量压缩应变(高达70%),而不发生破坏或开裂.玻璃纤维薄膜充当二氧化硅颗粒的支撑基体材料,显著提高了HSA 的力学性能.壳聚糖具有生物相容性、生物降解性、无毒性、化学活性好等特性,可作为一种良好的HSA 机械性能增强相[87].Ma 等[88]以TEOS 为硅源,壳聚糖为增强相,成功制备了壳聚糖-SiO2复合气凝胶,含20%(质量分数)壳聚糖的SiO2复合气凝胶具有高孔隙率(96.7%)、大孔隙体积(1.43 cm3·g-1)、大比表面积(618 m2·g-1)以及高水接触角(137°).此外,该气凝胶可以支撑自身重量7000 倍的物体.
引入有机聚合物可显著提高HSA 的机械性能,聚合物的引入拓宽了HSA 粒子间的颈部区域,提高了气凝胶结构的稳定性[89].Mahadik 等[90]采用高内相乳液(HIPE)工艺制备了柔性大孔聚合物-MTMS 基复合气凝胶.柔性大孔聚合物支撑了MTMS 基气凝胶的骨架结构,该气凝胶杨氏模量低至4.72×103N·m-2,呈现出极高的弹性、柔韧性以及超疏水性(165°).
5 疏水SiO2 气凝胶的吸油性能
HSA 不仅具有低密度、大比表面积和高孔隙率等特点,而且疏水/亲油性好,是一种很有应用前景的处理溢油污染的材料[4,91-93].
5.1 疏水SiO2 气凝胶吸油机理及吸油性能
HSA 吸油是化学吸附和物理吸附的结合,其中以物理吸附为主[94-95],化学吸附为辅.HSA 多孔结构引起的毛细管力是吸油的主要动力,其表面的亲油性烷基链可以和油类高分子发生溶剂化作用[96].在吸附过程中,油先扩散到气凝胶的外表面,然后迁移到孔隙中,在毛细管力或HSA 表面烷基链与油类高分子化学作用下,填充到气凝胶孔隙中.吸附能力主要受HSA 的孔隙率、疏水性、比表面积、表面能和溶剂表面张力等因素的影响[97].刘国强等[98]采用Langmuir 吸附等温方程描述了HSA 对亚甲基蓝(Methylene blue,MB)的吸附过程,吸附动力学符合准二级动力学方程,表明了吸附过程不是单一的,而是化学/物理复合吸附的过程.
Parale 等[99]采用两步酸碱催化溶胶-凝胶工艺制备了HSA 颗粒,研究了HSA 对四种烷烃、四种芳香化合物、四种醇和三种油的吸附,结果表明其对汽油吸附量为10.84 g·g-1,柴油11.56 g·g-1,发动机油12.91 g·g-1.Rao 等[4]以MTMS 为前驱体,利用溶胶-凝胶法结合超临界干燥,制备了柔性和高弹性的超疏水SiO2气凝胶.该气凝胶对油及有机物的吸附能力高达15 g·g-1左右,其中汽油13.82 g·g-1、煤油16.45 g·g-1以及柴油18.55 g·g-1,可作为一种用于石油泄漏清理的优异吸油材料.
Gurav 等[91]采用溶胶-凝胶法,结合常压干燥制备了低密度(0.065 g·cm-3)、超疏水性(153°)、高孔隙率(97%)的超疏水SiO2气凝胶.疏水改性降低了气凝胶的表面能,在毛细管力作用下,表面改性后的气凝胶表现出对油的高吸附能力,对汽油的吸附量为10.09 g·g-1,柴油为11.02 g·g-1.研究发现,被吸附的液体质量随着有机液体表面张力的增加而线性增加.
硅烷化剂所含官能团不同,吸附能力也有所不同.Çok 等[100]用单官能(TMCS)、三官能(MTMS、MTES)和有机官能硅烷(MEMO、GLYMO)为疏水改性剂,结合常压干燥制备了HSA 吸油材料.TMCS改性后的样品的孔隙率达到94%,密度低至0.13 g·cm-3,是油和有机液体良好的吸附剂,可以有效分离水中油或有机液体.TMCS 改性后的样品对煤油和柴油的吸附量分别达到12.5 g·g-1和11.6 g·g-1,而MEMO 改性后气凝胶的比表面积小、孔隙率和疏水性较低,导致其吸附能力较差.
常用改性剂制备的HSA 对油污和有机物的去除效率仍然很低,限制了其应用.为了提高SA 的吸油能力,Liu 等[101]以TEOS 为硅源,2,5-二乙烯基三甲氧基硅烷(DVTHP)为改性剂制备了超疏水噻吩桥联HSA.FTIR 和NMR 结果已经表明,HSA网络结构已经接上Si—CH3基团,并且当噻吩含量增加时,气凝胶疏水性增加.作为一种具有桥联链结构的新型有机硅烷,DVTHP 的加入会使得SA骨架结构孔径变大,提高其比表面积.当DVTHP与TEOS 的摩尔比为0.02∶1 时,该气凝胶比表面积达到834 m2·g-1,水接触角达到174°,噻吩单元的引入可以提高SA 吸附能力,对地沟油具有良好的吸附能力,其吸附量为16 g·g-1.
5.2 疏水SiO2 气凝胶吸附混合油
根据油水混合类型,可将油分为:游离油(油滴直径>150 μm)、分散油(油滴在20~150 μm 范围内)和乳化油(油滴<20 μm)[102-103],其中,乳化油最难处理.乳化油是油的一个重要存在形式[102],其含油浓度低,并且油滴无限期地保持乳化,难以去除.大多数气凝胶吸油材料只能去除水面或水下的油层,对表面活性剂稳定的水包油乳液的分离效果较差.HSA 具有超疏水性、高比表面积和超亲油性,使其能够分离油水混合物,即在油水混合物中,油相容易扩散到超亲油性的气凝胶孔隙中,HSA 表面的非极性基团和水相互排斥,从而实现油水分离[104].
Wang 等[105]以MTMS/十二烷基三甲氧基硅烷(DTMS)为前驱体,结合常压干燥制备了一种可以分离水中油层和水包油乳液的超疏水SiO2气凝胶.当DTMS 与MTMS 的质量比为4:6、硅烷浓度为18%时,气凝胶的密度低至0.087 g·cm-3,水接触角达到163°.同时也表现出最佳的吸附性能,即吸附能力达7.83~13.4 g·g-1,在氯仿中高达13.4 g·g-1,在煤油中为7.98 g·g-1,对于不同水包油乳液的分离效率可以达到98.4%以上.此外气凝胶重复使用16 次后,仍具有良好的吸附性能.
单一孔结构的HSA 在分离表面活性剂稳定的水包油乳液较为困难.Zhang 等[106]以MTES/TEOS为前驱体,在常压干燥条件下快速合成了小介孔(3~6 nm)和大介孔(17~30 nm)组成的双介孔HSA.大介孔可以吸附微米级的液滴,而小介孔对纳米级的液滴有很强的吸附性能,双介孔HSA 可用于油水分离.对正己烷、二氯甲烷、二甲基甲酰胺和乙醇的吸附能力达到8.4~17.1 g·g-1,能有效分离表面活性剂稳定的水包油乳液,分离效率高达97.6%.
原油是由烷烃、环烷烃和芳香烃等多种成分组成的复杂混合物,根据其密度可分为轻质原油和重质原油.Mahani 等[107]研究了MTMS 基HSA对轻质和重质原油的吸附性能.研究发现,吸附主要为分子间范德华力引起的物理吸附.在吸附过程中,HSA 的弹性以及其表面的甲基和原油分子间作用力,提高了吸附剂的吸附能力,其中对重质原油和轻质原油的吸附量分别达到16.7 g·g-1和13.7 g·g-1.
乳化油吸附与游离油的吸附机理不同.Wang等[108]以原油、植物油和机油为吸附质,研究了HSA 对纯液相油和油水乳状液中油的吸附机理.HSA 对纯液相油的吸油能力约为11.7~15.1 g·g-1,吸附速率依次为:原油>植物油>机油,而三种油的黏度则刚好相反,这表明气凝胶孔隙中的粘性流动为吸附的控速环节,并且吸附是毛细管力作用的物理吸附.此外,还采用了Freundlich 吸附模型拟合乳液相吸附等温线,用线性驱动力模型拟合了乳化液对油的吸附.在乳化油的吸附过程中,油滴首先迁移到气凝胶孔隙中,此为限制性环节,然后油在气凝胶孔表面进行吸附,吸附平衡仍然存在,但其动力学缓慢.亚微米级油滴的存在和表面活性剂的作用,使得乳液相吸附与溶液相吸附具有不同的机理.
5.3 疏水SiO2 气凝胶吸油再生性能
再生性是衡量HSA 吸油材料的关键指标[109].解吸是实现再生的关键步骤,根据气凝胶的柔韧性,其解吸过程可以分为两种:(1)对于柔韧性好的HSA,可以采用挤压的方法解吸[110];(2)对于脆性的HSA 则主要采用加热解吸,在加热解吸中,油分子蒸发过程分为两个过程:1)吸附质分子克服表面张力从孔隙内转移到气凝胶表面;2)根据吸附质的蒸汽压不同先后从气凝胶表面蒸发.表面张力越小,分子越容易到达表面,蒸发速度也就越快,解吸速率取决于相应有机液体的蒸汽压和表面张力[4].
HSA 的机械性能的增加,可以有效提高其再生性.Yun 等[111]以明胶和TEOS 为共前驱体,制备了明胶基HSA.明胶的加入增强了气凝胶的骨架结构,当添加质量分数为30%明胶时,气凝胶表现出优异的吸附能力和再生性,对油/有机溶剂吸附能力达到12~27 g·g-1,可重复使用10 次.Ma 等[88]制备的壳聚糖-SiO2复合气凝胶,对油和有机溶剂能力最高可达30 g·g-1,重复使用多达10 次后,油吸收能力几乎保持不变.周钰寒等[112]制备的纤维素基HSA 在50%范围内压缩后能完全恢复,机械性能好,吸油能力可达12.7 g·g-1.当气凝胶对原油的吸附达到饱和后,然后挤压解吸,经过10 次吸附/解吸循环后,仍具有90%的吸油能力.Shi 等[113]以聚丙烯腈纤维为增强相制备的HSA 可以有效吸附油水混合物中的油.添加质量分数为0.3%聚丙烯腈纤维的HSA 的机械性能最好,其压缩模量为260 kPa,聚丙烯腈在溶胶-凝胶过程前加入,并且均匀分散在二氧化硅基质中,不参与化学反应,只是起到支撑凝胶骨架的作用.采用浸没挤压法研究了该聚丙烯腈纤维HSA 的吸附/解吸循环性能,其对柴油的吸附能力达到9.56 g·g-1,经过100 次吸附/解吸后,仍能吸附柴油7.5 g·g-1,具有良好的再生性.
HSA 吸油是物理/化学吸附的并存的过程,其中物理吸附(毛细管力)占据主导地位.目前HSA吸附油的容量约为自身的十几倍,其吸附能力取决于吸附质的表面张力,吸附质的表面张力越大,吸附能力越强.HSA 具有超疏水性、高比表面积和超亲油性,能够高效率分离油水混合物,对水包油乳液的分离效率可以达到98%以上.HSA 机械性能差是影响其吸油再生性能的重要因素,经过机械性能增强后,可明显提高HSA 吸油材料的再生性.
6 结语与展望
SA 疏水改性主要采用表面后处理法和共前驱体法,这两种方法各有优缺点.目前,对表面后处理法-常压干燥制备HSA 的研究较多,而在共前驱体法制备HSA 方面有待深入.相比于超临界干燥,常压干燥工艺更具有实际生产应用的优势.HSA 的较大比表面积、高孔隙率、低密度以及疏水/亲油性使其可成为一种性能优异的吸油材料.然而,目前HSA 吸油材料制备也存在一些潜在的问题,需要进一步深入研究.
(1)开发低成本且环境友好的原料.合成HSA的原料主要有硅源、改性剂以及溶剂等,其昂贵的价格使得HSA 并未得到大规模工业化应用.目前常用的硅源主要是TMOS、TEOS、MTMS 等纯试剂,成本高.为了降低硅源成本,有研究采用相对廉价的水玻璃以及采用更为廉价的高岭土为原料制备HSA,但是对于采用高岭土或其他天然原料的研究还比较少,没有得到大范围的使用;无论是采用表面后处理法还是共前驱体法制备HSA,疏水改性剂主要为硅烷化剂(如TMCS、TMES、MTMS等),其成本高而且有些改性剂(如TMCS)污染环境,腐蚀设备,对其他疏水改性剂的研究还很少.因此,继续开发低成本且环境友好的原料,对于拓展HSA 的应用有重大意义.
(2)优化流程,开发周期短、易控制的疏水改性流程.表面后处理法和共前驱体法各有优劣,目前采用表面后处理法制备HSA 的研究较多,但是需要多次溶剂置换和多次改性处理,流程繁琐,有机溶剂和改性剂消耗量大,成本高,而且表面改性可能存在改性不彻底和疏水基团分布不均匀的问题.虽然有研究采用一步溶剂交换-表面改性法制备HSA,但其相关报道还很少,研究不系统,其制备工艺和相关反应机理有待进一步分析和论证;共前驱体法改性效果较好,但研究尚少,引入的疏水基团有限,并且其反应进程不好控制,有待深入研究.此外,对采用共前驱体法和表面后处理法组合改性工艺流程的研究同样较少.因此,需要进一步优化制备工艺,开发高效、周期短和易控制的疏水改性流程.
(3)大块体HSA 的制备仍然是严峻的挑战.常压干燥是目前制备HSA 的主要干燥工艺,但是一般只能制备小块HSA,并且需要结合多步流程(如选择表面张力低的溶剂置换、添加DCCA 等)才能达到优异的性能.为了在常压下制备出性能优异的大块体HSA,需要优化干燥工艺、开拓新的常压干燥工艺.
(4)HSA 的机械性能有待提高.目前已经采取了一系列的措施来增强HSA 的力学性能,如前驱体增强(采用含有机基团的前驱体)、物理增强(添加增强相)以及化学增强(引入聚合物)等,但是目前增强方法使用单一.为了更好的减少或避免单一方法带来的负面影响(优选有机基团的前驱体和物理增强对凝胶基体的增强程度有限;物理增强会增大HSA 的密度;化学增强会降低HSA 耐高温的性能),可重点研究多种增强方法协同使用,或者开拓新的增强途径,深入研究其内在的增强机理,从而达到更好的增强效果.
(5)HSA 吸油性能有待进一步提高.目前,HSA的吸油容量仅为自身的十几倍,相对于其他气凝胶吸油材料来说较低.大多数HSA 吸油材料只能去除水面或水下的油层,对于乳化油吸附及其机理研究却很少.所以加强HSA 吸附游离油、分散油和乳化油等多种油的机理研究是很有必要的.
(6)HSA 吸油材料的再生性有待提高.HSA 差的机械性能限制了其吸油再生性,因此提高再生性的关键是提高其机械性能.在目前的研究中,纤维增强的HSA 吸油材料的再生性较好,其循环使用次数可达数十次.对于其他复合材料的增强行为及其再生性的研究还比较少.因此,继续开展多种强化方法协同使用,或者开拓新的增强途径,对强化HSA 机械性能进而提高HSA 吸油材料再生性有重要意义.
随着研究的深入和工艺的不断优化,HSA 必将朝着成本低、疏水性好、力学性能好、吸附能力强和再生性能好的方向发展,并且能够在吸油领域上实现大规模实际应用.
猜你喜欢
中国特种设备安全(2021年5期)2021-11-06
云南化工(2020年11期)2021-01-14
陶瓷学报(2020年5期)2020-11-09
纺织科学与工程学报(2020年1期)2020-06-12
中国生物医学工程学报(2019年4期)2019-07-16
船海工程(2019年3期)2019-07-03
材料科学与工程学报(2016年4期)2017-01-15
中国塑料(2015年5期)2015-10-14
上海塑料(2015年3期)2015-02-28
油气田环境保护(2014年3期)2014-04-27
