
玉米穗长一般配合力多位点全基因组关联分析和预测
2023-04-12 01:38朱卫红刘京宝郭国俊
作物学报 2023年6期
马 娟 朱卫红 刘京宝 宇 婷 黄 璐 郭国俊
玉米穗长一般配合力多位点全基因组关联分析和预测
马 娟*朱卫红 刘京宝 宇 婷 黄 璐 郭国俊
河南省农业科学院粮食作物研究所, 河南郑州 450002
穗长是一个重要的农艺性状, 与产量密切相关。一般配合力(general combining ability, GCA)是评价优异自交系的重要指标。因此, 解析穗长GCA的遗传基础, 制定相应的育种策略对玉米杂交种产量的提高具有重要意义。本研究以123个玉米自交系和8个测验种按照North Carolina II遗传交配设计组配的537个F1杂交种为试验材料, 在2个环境下进行表型鉴定, 利用玉米5.5 K液相育种芯片鉴定的11,734个SNP (single nucleotide polymorphisms)对2个环境以及综合环境穗长GCA进行多位点全基因组关联分析(multi-locus genome-wide association study, MGWAS)和基因组预测。利用7种MGWAS共检测到11个穗长GCA显著关联SNP标记(< 8.52E-07), 单个位点解释GCA变异介于8.06%~28.23%之间。不同MGWAS共定位的SNP位点有5个。位点7_178103602在周口和综合环境利用mrMLM (multi-locus random-SNP-effect mixed linear model)方法重复检测到, 可解释穗长GCA变异的26.02%~28.23%, 为环境稳定的主效SNP。共挖掘10个候选基因, 其中和EID1-like F-box protein 2可能是控制穗长GCA的关键基因。5种随机效应模型对3个环境穗长GCA的预测准确性介于0.53~0.69之间, 且模型间差异较小。在新乡和周口环境, GBLUP (genomic best linear unbiased prediction)和RKHS (reproducing kernel Hilbert space)整合不同显著位点作为固定效应均可提高穗长GCA基因组估计育种值的准确性, 提高率为2.34%~14.98%, 而在综合环境中除了利用FarmCPU (fixed and random model circulating probability unification)或BLINK (Bayesian-information and linkage-disequilibrium iteratively nested keyway)鉴定的1个显著位点作为固定效应会略降低预测精度外, 其他2种MGWAS方法显著位点的加入均能提高基因组预测力, 提高率为2.80%~6.84%。因此, MGWAS显著位点作为固定效应加入预测模型有利于提高穗长GCA基因组估计育种值的准确性, 可用来对玉米亲本穗长GCA进行有效预测和选择。
穗长; 一般配合力; 多位点全基因组关联分析; 固定效应模型; 基因组选择
玉米是杂种优势利用最典型的作物之一, 主要体现在单交种的生产和利用上。自交系配合力鉴定和评价是玉米杂交育种的一个重要环节。配合力包括一般配合力(general combining ability, GCA)和特殊配合力(special combining ability, SCA)。GCA是评价一个自交系与其他自交系杂交后代在某个性状上的平均表现, 而SCA是评价杂交组合组配优劣的参考指标。相比SCA, GCA由加性效应控制, 可以稳定遗传。因此, 研究GCA的遗传机制对选育高配合力的亲本材料更具有实际应用价值。直观上玉米果穗长度即穗长决定行粒数的多少, 而行粒数是产量构成因子穗粒数的决定因子之一。通过基因克隆, 张人予[1]发现穗长基因在不显著改变玉米株型和穗型的基础上, 可以显著增加穗粒数; Jia等[2]发现一个编码丝氨酸/苏氨酸蛋白激酶的行粒数基因调控雌蕊小花数和穗长。相比单株或小区产量,穗长的遗传力较高, 可作为间接选择性状。因此, 揭示穗长GCA的遗传机制对玉米产量遗传改良具有重要意义。
连锁分析和全基因组关联分析(genome-wide association study, GWAS)是挖掘穗长GCA关键位点和解析其遗传机理的重要方法。周广飞[3]利用玉米F2:3家系和其测交群体在7号染色体鉴定到6个控制穗长GCA效应的遗传位点, 其中5个也是控制穗长本身的遗传位点。Liu等[4]利用194个重组自交系与郑58和B73、HD568和Mo17以及所有4个测验种组配的3个North Carolina II (NCII)杂交群体挖掘到13个控制穗长GCA的QTLs (quantitative trait loci), 其中位于4号染色体上(35.76~62.54 Mb)的1个主效QTL在3个杂交群体中均检测到。目前, 利用单位点GWAS方法对穗长GCA开展了全基因组鉴定研究。监立强[5]以248份玉米自交系和其组配的400个F1杂交组合为试验材料, 利用MLM (mixed linear model)的Q (群体结构) + K (亲缘关系)模型挖掘到13个穗长GCA显著位点, 其变异解释率均大于10%。刘文童等[6]利用单位点方法SUPER (settlement of MLMs under progressively exclusive relationship)检测到3个控制玉米穗长GCA的显著关联位点, 其解释GCA效应的变异率较低, 为0.01%~4.34%, 赖氨酸和组氨酸特异性转运体为穗长GCA的候选基因。监立强[5]和刘文童等[6]研究中没有找到穗长本身和GCA一致的SNPs位点, 表明GCA的遗传基础与性状本身的遗传基础可能存在差异。以上研究明确了穗长GCA效应数量性状遗传的本质, 为深入剖析其遗传基础提供了丰富的遗传学信息。
由于穗长GCA是数量性状, 受多基因控制, 因此采用单位点模型对其进行遗传解析具有一定的局限性。多位点GWAS (multi-locus GWAS, MGWAS)方法考虑相邻位点的潜在关系[7], 比单位点模型更能解释多基因性状的遗传基础。针对单位点模型的局限性, 大量多位点GWAS模型被开发。为了解决群体结构、亲缘关系和候选标记间的混杂问题, Liu等[8]提出了一种交替使用固定效应模型和随机效应模型的多位点GWAS模型即FarmCPU (fixed and random model circulating probability unification), 其中固定效应模型利用最大似然法选择潜在关联位点,而随机效应模型则利用bin方法预测优化显著关联位点。相比FarmCPU算法, BLINK (linkage-disequilibrium iteratively nested keyway)不仅利用连锁不平衡代替bin方法提高统计功效, 还利用固定效应模型的贝叶斯信息指标代替随机效应模型中的最大似然法来提高运算速度[9]。mrMLM (multi-locus random-SNP-effect mixed linear model)首先利用单标记扫描策略, 根据宽松的阈值选定潜在关联SNP, 其次将潜在SNP拟合进多位点模型, 通过经验贝叶斯估计和似然比测验最终获得显著关联的SNP[10]。为了提高mrMLM方法的运算速度, Tamba和Zhang[11]提出了FASTmrMLM (FAST multi-locus random-SNP-effect mixed linear model), 其核心是基于全基因组有效混合模型关联算法、矩阵转化和等式检测显著标记。FASTmrEMMA (FAST multi-locus random-SNP-effect EMMA)的原理是将SNPs作为随机效应, 并对多基因矩阵K和环境噪声协方差矩阵进行漂白处理, 并指定非零特征值的个数等于1[12]。pLARmEB (polygene-background-control-based least angle regression plus empirical Bayes)主要对FASTmrEMMA算法中的矩阵变换进行拓展[13]。ISIS EM-BLASSO (iterative sure independence screening EM-Bayesian LASSO)是利用安全独立筛选-平滑削边绝对偏差惩罚(SIS-SCAD)算法筛选潜在SNPs标记, EM-BLASSO算法和似然比测验获得显著SNP[14]。目前, 多位点GWAS被广泛应用于玉米等作物重要农艺性状的遗传研究中[15-19], 但对玉米穗长GCA的研究鲜见报道。
尽管连锁和关联分析研究揭示了控制数量性状的关键位点和候选基因, 但如何利用这些关键位点以及针对目标性状制定相应的分子育种策略仍是困扰育种工作者的一个关键问题。基因组选择(genomic selection, GS)是目前主流的一种分子育种技术, 最早由Meuwissen等[20]提出。GS能够利用影响性状的所有变异位点估计育种值并进行有效选择, 可通过缩短育种周期来大幅度提高遗传进度。为提高GS的准确性, 研究者发展了很多统计模型和算法例如基因组最佳线性无偏预测(genomic best linear unbiased prediction, GBLUP)、贝叶斯模型和非参数模型等[20-23], 其目的均是通过有效降维从而实现对标记效应的准确估计。目前, GS广泛应用于玉米等作物亲本自交系和杂交种产量等性状表型预测[24-27]。而且, 利用GS对亲本材料产量性状GCA效应也开展了相关预测研究。例如, Wang等[28]通过模拟和真实数据提出利用稀疏部分双列杂交设计可以有效预测玉米单穗重的GCA效应。Zhang等[29]利用32个玉米自交系和9个测验种组配的3组杂交种数据对产量GCA效应进行了预测。研究发现, 自交系和测验种联合预测的效果最好。目前利用GS策略对穗长GCA进行基因组预测研究尚未见报道。
为进一步解析玉米穗长GCA的遗传机制, 提出有效的分子育种改良策略, 本研究选用123个玉米自交系和8个测验种按照NCII遗传交配设计获得的537个F1杂交组合为试验材料, 采用7种MGWAS方法挖掘穗长GCA显著关联位点, 并利用GBLUP和RKHS (reproducing kernel Hilbert space)研究显著关联位点作为固定效应对提高穗长GCA预测精度的影响。
1 材料与方法
1.1 试验材料、田间设计及配合力分析
根据NCII遗传交配设计, 利用123个玉米自选系与8个测验种(M189、M119、20H1419、S110T、L119A、PH4CV、昌7-2和农系531)组配了537个F1杂交种。测验种昌7-2、PH4CV、M189和农系531分别是玉米杂交品种郑单958的父本、先玉335的父本、郑单309的父本和农单5316的母本, 其他4个为自选系。537个F1杂交子代于2021年种植在河南新乡和周口试验田。采用随机区组试验设计, 两次重复, 每个材料每小区均种植1行。新乡试验田小区行长、株距和行距分别是4.00、0.22和0.60 m, 而周口试验田小区行长、株距和行距分别是3.30、0.22和0.60 m。收获后, 每小区每个材料选取单穗测量穗长(cm)。根据537个F1杂交子代穗长表型, 利用R语言lme4包分别计算新乡和周口环境131个玉米自交系穗长的GCA效应值, 同时将两环境联合计算,获得综合环境穗长GCA值。根据下面公式计算穗长GCA的遗传力:

1.2 基因型鉴定及分析
利用河南省农业科学院粮食作物研究所开发的玉米5.5 K液相育种芯片(5521个靶向探针位点)对131个玉米自交系进行基因型鉴定, 测序平台为Illumina NovaSeq 6000。利用BWA软件将过滤的reads与玉米B73第4版参考基因组(http://www. gramene.org/)进行比对。利用GATK v4.1.2.0软件检测到33,971个原始SNPs标记。将最小等位基因频率<0.05, 缺失率>10%和杂合率>1%的SNPs标记过滤后, 共获得11,734个SNP用于后续关联分析和基因组预测分析。
1.3 显著位点和候选基因的挖掘
多数研究表明, 相比只考虑群体结构Q或是亲缘关系K的GWAS模型, 同时考虑两者的GWAS模型具有较好的拟合性[30-32]。因此, 本研究利用7种MGWAS方法(BLINK、FarmCPU、FASTmrMLM、FASTmrEMMA、mrMLM、pLARmEB和ISIS EM- BLASSO)的Q+K模型对新乡、周口和综合环境穗长GCA进行关联分析。利用Structure v2.3.4[33]软件计算群体结构Q值, 其中亚群数设置为1~10, Burnin期的长度设置为5000, 蒙特卡罗重复个数设为50,000, 每个亚群数重复3次。根据Δ的结果, 确定最佳的亚群数为6。根据Structure软件结果, 利用CLUMPP软件的FullSearch方法[34]获得最终值。亲缘关系值采用TASSEL v5.0[35]软件的Centered_ IBS算法确定。除了BLINK和FarmCPU利用GAPIT R包[36]计算, 其他方法均采用mrMLM R包[37]计算。利用< 8.52E-07 (0.01/11,734)确定标记与穗长GCA关联的显著性。利用SnpEff[38]对显著位点的候选基因信息进行挖掘, 参数按默认设置。利用MaizeGDB数据库中qTeller工具获取预测基因以及3个已知穗长基因([2]、[39]和[40])在B73等自交系不同组织的表达量。根据表达量, 利用R语言hclust函数对候选基因和已知基因进行聚类, 方法为最长距离法。通过R语言的GENIE3包的随机森林算法推测基因之间的互作网络, 互作关系权重阈值设置为0.1。基因间的网络关系采用Cytoscape v3.9.1展示。
1.4 基因组选择模型和策略
利用Bayes A、Bayes C、贝叶斯最小绝对缩减和变量选择算子(Bayesian least absolute shrinkage and selection operator, Bayesian LASSO)、GBLUP和RHKS多核模型对2个环境以及综合环境穗长GCA进行基因组预测分析。5种预测方法中所有标记的效应均为随机效应, 定义该模型为随机效应模型。为研究MGWAS显著位点对GCA预测精度的影响, 本研究构建了GBLUP和RKHS预测方法的固定效应模型。在固定效应模型中, 根据不同MGWAS方法挖掘的关联位点信息, 将显著SNPs标记设定为固定效应, 其余标记设定为随机效应。采用10倍交叉验证方式将131个自交系分为训练集和验证集, 重复100次。评价不同模型的指标为验证集基因组估计育种值与表型值的相关系数均值。所有模型和方案均在BGLR R包中实现[41], 其中蒙特卡洛马尔科夫链长为12,000, 预烧为3000, 其他参数按默认设置。预测准确性方差分析和Duncan多重比较均采用R语言计算。
2 结果与分析
2.1 穗长一般配合力效应值统计结果
新乡、周口和综合环境穗长GCA效应值分别介于1.45~1.95 cm、0.71~0.82 cm和1.24~1.33 cm (图1-A)。新乡和周口环境之间表现为中度正相关关系(= 0.57), 均与综合环境高度正相关(= 0.87~0.89) (图1-B)。方差分析表明, 自交系和测验种的GCA、SCA、GCA或SCA与环境互作效应均达到显著或极显著水平(表1)。穗长GCA的遗传力较高, 为0.82。这些结果表明, 虽然穗长GCA主要受遗传因素控制, 但同时也受到环境因素的影响。
2.2 穗长一般配合力显著关联位点和候选基因
利用7种MGWAS方法共检测到11个穗长GCA显著关联SNP (< 8.52E-07), 其解释GCA变异率介于8.06%~28.23%之间(表2)。新乡、周口和综合环境均检测到4个显著关联位点。其中, 位点7_178103602在周口和综合环境利用mrMLM方法重复检测到, 其解释穗长GCA变异率为26.02%~ 28.23%, 为环境稳定的主效SNP。不同MGWAS方法共定位的SNP位点有5个。在新乡环境, 位点2_216138581和8_126983650利用FASTmrEMMA和pLARmEB均检测到, 其能解释穗长GCA表型变异的15.63%~23.09%, 为控制穗长GCA的主效位点。7_178327031为FASTmrMLM和pLARmEB方法共定位的SNP, 可以解释周口环境穗长GCA变异的10.64%~11.29%, 也是控制穗长GCA的主效位点。
利用SnpEff共挖掘到穗长GCA候选基因10个, 有注释信息的基因为5个(表2)。环境稳定的主效位点7_178103602挖掘的候选基因为编码过氧化物酶的()。不同MGWAS方法共定位的2个主效位点7_178327031和2_216138581对应的候选基因分别为编码生长素氨基合成酶的()和编码类EID1 F-框蛋白的EID1-like F-box protein 2 (EDL2)。利用MaizeGDB数据库中qTeller工具获得了10个候选基因以及3个已知穗长基因(、和)在B73等自交系不同组织的表达量(附表1)。相比其他组织,和ELD2分别在胚和成熟雌穗小花中高表达(附表1)。聚类结果表明,与的欧式距离较近, 两者可能具有相似的表达模式; ELD2也与划分为一大类(图2-A)。GENIE3预测的基因互作网络结果表明, ELD2与、可能存在互作关系, 而与、相互关联(图2-B)。
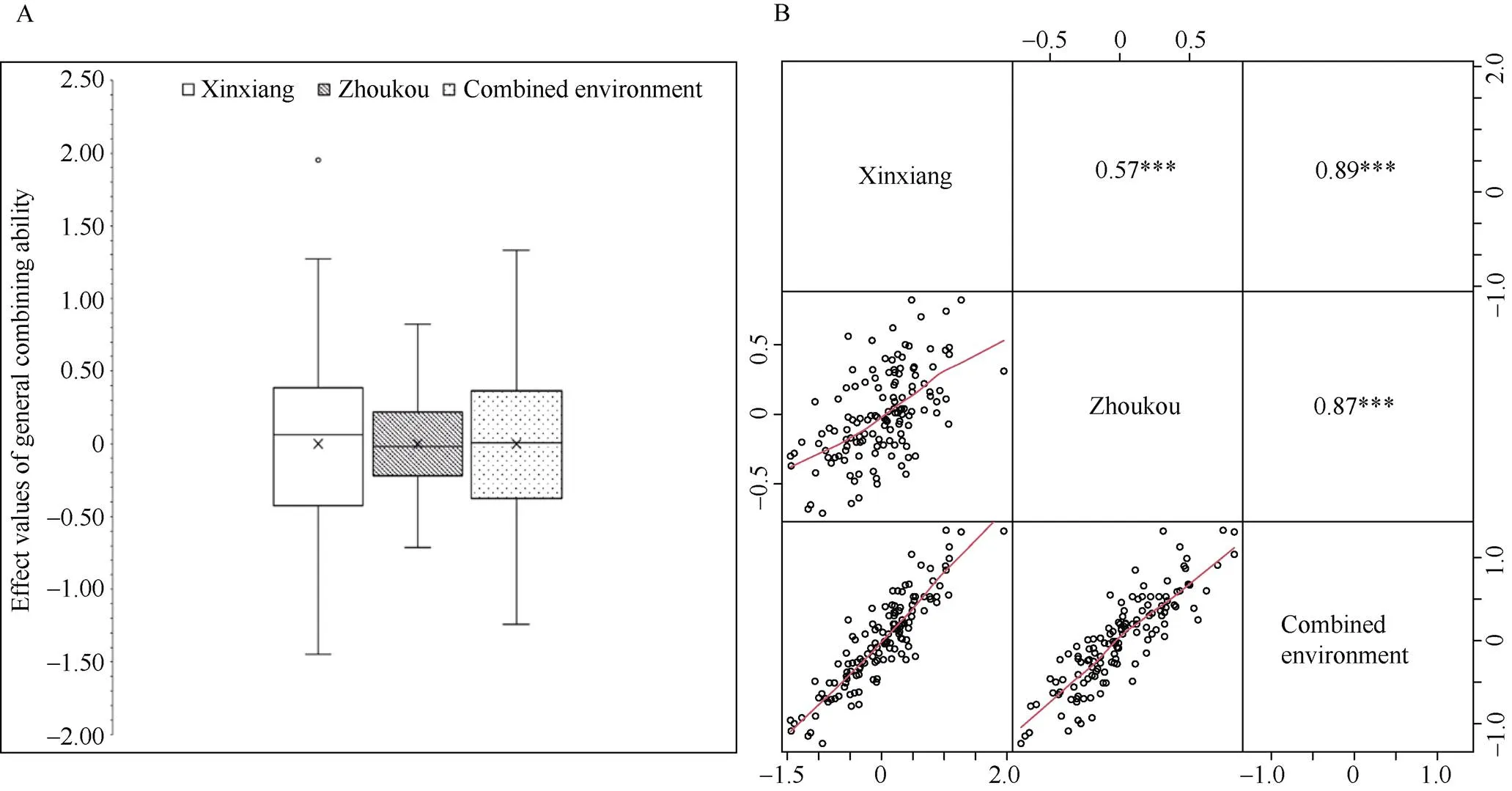
图1 穗长一般配合力分布(A)和不同环境相关性(B)
图A中×表示均值。图B中***表示在0.001概率水平差异显著。
In Fig. 1-A, × represents means. In Fig. 1-B, *** represents there is significant difference at the 0.001 probability level.

表1 方差分析和穗长一般配合力的遗传力
P1和P2分别表示自交系和测验种。
P1 and P2 represent the inbred lines and tester lines, respectively.

表2 穗长一般配合力显著关联SNP和候选基因
2为单个位点表型变异解释率。2represents phenotypic variance explained by one locus.
2.3 不同随机效应模型的预测准确性
利用5种GS随机效应模型对新乡、周口和综合环境穗长GCA进行了基因组预测分析。方差分析和Duncan多重比较分析表明, 3个环境之间的预测准确性差异显著(附表2和附表3)。综合环境穗长GCA的预测准确性最高, 为0.67~0.69, 其次为新乡环境(0.60~0.61), 周口环境的准确性最低, 为0.53~0.54 (图3)。5种随机效应模型对穗长GCA效应估计育种值的准确性差异较小且不显著(附表2), 最大差值仅为0.02。3个环境中, Bayesian LASSO的基因组预测能力均最低, 而其他4个模型的预测准确性基本相等。

图3 5种随机效应模型对穗长一般配合力的预测准确性
2.4 多位点全基因组关联分析先验信息对预测准确性的影响
考虑到模型之间的差异较小以及Bayes模型的运算时间较长, 本研究仅利用GBLUP和RKHS方法研究了7种MGWAS方法挖掘的显著SNP位点作为固定效应对提高穗长GCA基因组预测力的影响。除了新乡环境外, 周口和综合环境不同模型之间预测准确性存在显著差异(附表4和附表5)。对于新乡和周口环境, GBLUP和RKHS整合不同显著位点作为固定效应均可提高穗长GCA基因组估计育种值的准确性, 提高率为2.34%~14.98% (图4-A, B)。在综合环境中, 利用FarmCPU或BLINK鉴定的1个显著位点作为固定效应会略降低预测的准确性, 其他2种MGWAS方法均能提高预测力, 提高率为2.80%~6.84% (图4-C)。
3 讨论
3.1 显著阈值和多位点GWAS模型的选择
近年来, MGWAS方法在植物遗传研究中得到广泛应用, 其在多重检验、群体结构和多基因背景控制方面的优势也逐渐凸显出来[7,42]。在MGWAS方法中, 所有潜在关联标记和效应能够在一个线性模型中同时确定并估计出来, 因此无需进行Bonferroni校正[7,42]。mrMLM软件包的开发者建议将LOD = 3 (= 0.0002)作为多位点模型显著位点的临界值。由于GCA效应值有正负之分, 相比性状本身, 其变异较大, 因此本研究仍选择了0.01水平下的Bonferroni矫正来控制假阳性率。尽管设置了严格的阈值, 本研究利用7种多位点模型仍检测到11个控制穗长GCA的显著关联位点。这些结果表明, 多位点模型在检测GCA关键位点方面具有较高的检测功效。
本研究中pLARmEB方法对穗长GCA的检测功效最高, FASTmrMLM最保守。Yang等[15]发现pLARmEB对小麦籽粒品质性状和面团流变特性的检测功效高于mrMLM和FASTmrEMMA, 但低于FASTmrMLM。对于水稻耐盐性多位点GWAS方法研究, pLARmEB的检测功效次于ISIS-BLASSO和FASTmrMLM, 但高于mrMLM和FASTmrEMMA[18]。Zhou等[19]采用6种多位点GWAS方法对玉米成熟期籽粒含水量进行定位研究得出, ISIS-BLASSO检测的位点个数最多, pLARmEB方法相对保守。An等[43]发现在多个环境中mrMLM模型对玉米穗行数均具有最高的检测功效, pLARmEB居中, FASTmrEMMA最保守。由于不同多位点GWAS方法在统计模型、潜在关联位点选择策略以及显著位点检验方法等方面的差异, 可能导致了其对不同性状检测功效的不同。多数研究表明, 利用多种GWAS方法有助于挖掘稳定的变异位点[15-19]。本研究发现不同模型共定位的位点有5个, 而且多为控制穗长GCA的主效位点。因此, 利用多种MGWAS方法有助于提高位点检测的可靠性, 为后续功能验证提供可靠的基因信息。

图4 GBLUP和RKHS整合显著位点作为固定效应的预测准确性
A、B和C分别表示新乡、周口和综合环境。GBLUP和RKHS表示随机效应模型。GBLUP和RKHS作为后缀的模型表示固定效应模型。
A, B, and C represent Xinxiang, Zhoukou, and combined environment, respectively. GBLUP and RKHS represent random effect models. Models with GBLUP and RKHS as suffixes represent fixed effect models.
3.2 不同定位研究结果比较
本研究中绝大多数位点对穗长GCA变异的解释率大于10%, 这与监立强[5]利用单位点混合线性模型得出的研究结果一致。检测的9个显著位点与前人利用双亲群体定位的穗长、行粒数、产量和单株产量QTL或MQTL存在重叠。位点1_192956360同时被BLINK和FarmCPU方法检测到, 其位于Zhou等[44]利用四交群体定位的一个控制穗长的QTL置信区间内。位于7号染色体上的3个显著位点均位于一个测交群体定位的单株产量QTL[45]和一个对产量和单穗重具有多效性的QTL区间内[46]。3个位点(6_84476172、7_106761824、8_126983650)均位于Chen等[47]利用Meta-QTL整合的产量、穗部性状和籽粒性状的MQTL内。位点10_149653708位于一个控制行粒数和穗行数的MQTL区间内[48]。位点5_194917385与一个同时控制产量、单穗重的QTL和一个控制小区产量GCA的QTL存在重叠[49-50]。不同群体间重叠的基因组区域说明影响穗长GCA的关联位点可能对性状本身、产量以及行粒数具有多效性。
3.3 穗长一般配合力候选基因预测
生长素氨基合成酶和类EID1 F-框蛋白EDL2是不同MGWAS方法共定位主效位点预测的候选基因。编码生长素/吲哚乙酸蛋白的基因BARREN INFLORESCENCE1和BARREN INFLORESCENCE4是生长素信号途径调控玉米花序形成不可或缺的关键因子[51], 说明生长素在玉米穗部小花的发育调控制中具有重要作用。已知穗长和行粒数基因编码丝氨酸/苏氨酸蛋白激酶, 通过介导Arf GTPase-activating protein的磷酸化来调控生长素依赖的花序发育, 从而影响玉米穗长和产量[2]。玉米穗长基因通过参与糖和生长素信号途径影响玉米雌穗花序分生组织的发育, 进而影响玉米穗长和单穗产量[39]。本研究利用一种随机森林集成算法发现与、可能存在互作关系。拟南芥中, 过表达和条件敲除突变体研究发现EDL3是脱落酸依赖的信号级联反应的正向调节因子, 而脱落酸信号传导途径在控制种子萌发、开花转换等发育过程起着重要作用[52]。穗长基因通过控制玉米花序中内源乙烯生物合成水平影响小花败育率, 从而调节玉米穗长和穗粒数。玉米EDL2是拟南芥EDL3的同源子, 其与穗长基因、存在互作。而且,和EDL2分别在胚和成熟雌穗小花中高表达(附表1)。因此,和EDL2可能是调控玉米穗长GCA的关键基因。
3.4 穗长一般配合力全基因组预测策略
本研究中5种随机效应模型对玉米穗长GCA具有相似的基因组预测力, 与玉米产量及产量相关性以及小麦黄锈病预测研究结论一致[53-54]。由于穗长GCA的遗传力较高, 利用5种随机效应模型均取得了中等及以上的预测准确性。但相比遗传力, 穗长GCA的预测精度仍有提高的空间。根据MGWAS定位的结果, 本研究将1~2个显著位点作为固定效应加入GBLUP和RKHS模型对穗长GCA效应开展了基因组预测分析。结果发现, 相比随机效应模型, 除了综合环境FarmCPU或BLINK方法挖掘的1个显著位点作为固定效应不能提高预测精度外, 其余情况均能提高GCA基因组估计育种值的准确性, 提高率为2.34%~14.98%。Ma和Cao[55]将单位点模型CMLM和多位点模型FarmCPU挖掘的4~8个控制玉米穗行数和穗长的显著SNPs作为固定效应整合到5种预测模型(GBLUP、RKHS、Bayes A、Bayes B和Bayes C)中也得到了相同的结论。此外, 将主效的生长习性基因作为固定效应整合到预测模型也能提高小麦产量的预测准确性[56]。Odilbekov等[57]发现将3个单位点模型(GLM、MLM和SUPER)和2个多位点模型(FarmCPU和MLMM)检测的1~3个显著关联位点作为固定效应能够提高小麦叶枯病的预测力。将连锁分析定位的显著QTL作为固定效应也可提高性状本身的预测准确性。在玉米BC3F5群体中, 相比完全随机效应模型, 将1~2个QTL作为固定效应整合到6种预测模型中可以提高穗行数、穗粒数、行粒数和叶夹角基因组估计育种值的准确性[58]。Arruda等[59]发现将控制小麦赤霉病的QTL作为固定效应拟合到ridge regression BLUP (RR-BLUP)模型中能够提高小麦赤霉病的基因组预测力。通过模拟, Bernardo[60]认为将表型变异解释率大于10%的主效基因作为固定效应加入RR-BLUP模型总会提高性状的预测力。本研究中除了BLINK和FarmCPU检测的位点外, 多数位点对GCA变异的解释率大于10%, 这可能是固定效应模型可以提高穗长GCA预测力的原因之一。
此外, 研究者还发现相比随机选择的位点, 将GWAS挖掘的显著位点全部作为随机效应也可以提高性状本身的预测准确性。对于干旱和高温环境的玉米产量和开花期, RR-BLUP利用8~339个显著关联的SNP获得的预测准确性高于随机选择的覆盖全基因组的10,108个SNP[61]。在热带玉米种质中, Liu等[62]发现仅利用GWAS显著关联位点作为标记集对玉米茎腐病的准确性高于覆盖基因组的所有标记。在小麦中, Cericola等[63]也认为相比随机选择的标记, GWAS衍生的标记有助于提高产量、倒伏和淀粉含量的预测准确性。因此, GS整合MGWAS挖掘的性状关联标记可以提高GCA全基因组预测的准确性, 可以用来预测和选择穗长配合力较高的亲本材料。
4 结论
本研究利用7种MGWAS共检测到11个控制穗长GCA的显著关联SNP, 不同MGWAS共定位的SNP有5个, 环境稳定的有1个。生长素氨基合成酶和类EID1 F-框蛋白ELD2可能是控制穗长GCA的关键基因。5种随机效应模型对3个环境穗长GCA的预测准确性介于0.53~0.69之间, 可以有效预测穗长GCA效应值。将本研究检测到的MGWAS显著位点作为固定效应加入预测模型有利于提高穗长GCA基因组估计育种值的准确性。
附表 请见网络版: 1) 本刊网站http://zwxb. chinacrops.org/; 2) 中国知网http://www.cnki.net/; 3) 万方数据http://c.wanfangdata.com.cn/Periodical- zuowxb.aspx。
[1] 张人予. 玉米穗长基因EL3的克隆及我国优良自交系基因组变异分析. 中国农业大学博士学位论文, 北京, 2018. Zhang R Y. Cloning of EL3 for Ear Length in Maize and Patterns of Genomic Variation in Chinese Maize Inbred Lines. PhD Dissertation of China Agricultural University, Beijing, China, 2018 (in Chinese with English abstract).
[2] Jia H T, Li M F, Li W Y, Liu L, Jian Y N, Yang Z X, Shen X M, Ning Q, Du Y F, Zhao R, Jackson D, Yang X H, Zhang Z X. A serine/threonine protein kinase encoding generegulates maize grain yield., 2020, 11: 988–998.
[3] 周广飞. 一个控制玉米行粒数、穗长其一般配合力的多效性QTL(qKNR7.2)鉴定. 华中农业大学硕士学位论文, 湖北武汉,2014. Zhou G F. Identification of A Pleitropic QTL (qKNR7.2) for Kernel Row Number Per Row, Ear Length, and General Combining Ability of Maize. MS Thesis of Huazhong Agricultural University, Wuhan, Hubei, China, 2014 (in Chinese with English abstract).
[4] Liu X G, Hu X X, Li K, Liu Z F, Wu Y J, Feng G, Huang C L, Wang H W. Identifying quantitative trait loci for the general combining ability of yield-relevant traits in maize., 2021, 71: 217–228.
[5] 监立强. 玉米产量相关性状及其一般配合力的关联分析. 河北农业大学硕士学位论文, 河北保定, 2017. Jian L Q. Genome-Wide Association Study of Yield-Related Traits and General Combining Ability in Maize (L.). MS Thesis of Hebei Agricultural University, Baoding, Hebei, China, 2017 (in Chinese with English abstract).
[6] 刘文童, 监立强, 郭晋杰, 赵永锋, 黄亚群, 陈景堂, 祝丽英. 玉米穗部性状及其一般配合力的关联分析. 植物遗传资源学报, 2020, 21: 706–715. Liu W T, Jian L Q, Guo J J, Zhao Y F, Huang Y Q, Chen J C, Zhu L Y. Association analysis of ear-related traits and their general combining ability in maize., 2020, 21: 706–715 (in Chinese with English abstract).
[7] 温阳俊, 冯建英, 张瑾. 多位点关联分析方法学的研究进展. 南京农业大学学报, 2022, 45: 1–10. Wen Y J, Feng J, Zhang J. Research progress of mulit-locus genome-wide association study., 2022, 45: 1–10 (in Chinese with English abstract).
[8] Liu X L, Huang M, Fan B, Buckler ES, Zhang Z. Iterative usage of fixed and random effect models for powerful and efficient genome wide association studies., 2016, 12: e1005767.
[9] Huang M, Liu X, Zhou Y, Summers RM, Zhang Z W. BLINK: a package for the next level of genome-wide association studies with both individuals and markers in the millions., 2019, 8: 1–12.
[10] Wang SB, Feng JY, Ren WL, Huang B, Zhou L, Wen YJ, Zhang J, Dunwell JM, Xu S, Zhang YM. Improving power and accuracy of genome-wide association studies via a multi-locus mixed linear model methodology., 2016, 6: 19444–19453.
[11] Tamba CL, Zhang YM. A fast mrMLM algorithm for multi-locus genome-wide association studies., 2018. https://doi.org/10.1101/341784.
[12] Wen YJ, Zhang H, Ni YL, Huang B, Zhang J, Feng JY, Wang SB, Dunwell JM, Zhang YM, Wu R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies., 2018, 19: 700–712.
[13] Zhang J, Feng JY, Ni YL, Wen YJ, Niu Y, Tamba CL, Yue C, Song Q, Zhang YM. pLARmEB: integration of least angle regression with empirical Bayes for multilocus genome-wide association studies., 2017, 118: 517–524.
[14] Tamba CL, Ni YL, Zhang YM. Iterative sure independence screening EM-Bayesian LASSO algorithm for multi-locus genome-wide association studies., 2017, 13: e1005357.
[15] Yang Y, Chai Y M, Zhang X, Lu S, Zhao Z C, Wei D, Chen L, Hu YG. Multi-locus GWAS of quality traits in bread wheat: mining more candidate genes and possible regulatory network., 2020, 11: 1091–1109.
[16] Peng Y C, Liu H B, Chen J, Shi T T, Zhang C, Sun D F, He Z H, Hao Y F, Chen W. Genome-wide association studies of free amino acid levels by six multi-locus models in bread wheat., 2018, 9: 1196–1204.
[17] Su J J, Wang C X, Hao F S, Ma Q, Wang J, Li J L, Ning X Z. Genetic detection of lint percentage applying single-locus and multi-locus genome-wide association studies in Chinese early-maturity upland cotton., 2019, 10: 964–974.
[18] Cui Y R, Zhang F, Zhou Y L. The application of multi-locus GWAS for the detection of salt-tolerance loci in rice., 2018, 9: 1464–1472.
[19] Zhou G F, Zhu Q L, Mao Y X, Chen G Q, Xue L, Lu H H, Shi M L, Zhang Z L, Song X D, Zhang H M, Hao D R. Multi-locus genome-wide association study and genomic selection of kernel moisture content at the harvest stage in maize., 2021, 12: 697688–697700.
[20] Meuwissen T H, Hayes B J, Goddard M E. Prediction of total genetic value using genome-wide dense marker maps., 2001, 157: 1819–1829.
[21] Vanraden P M. Efficient methods to compute genomic predictions., 2008, 91: 4414–4423.
[22] de los Campos G, Naya H, Gianola D, Crossa J, Legarra A, Manfredi E, Weigel K, Cotes J M. Predicting quantitative traits with regression models for dense molecular markers and pedigree., 2009, 182: 375–385.
[23] González-Recio O, Forni S. Genome-wide prediction of discrete traits using bayesian regressions and machine learning., 2011, 43: 7–18.
[24] Guo Z G, Tucker D M, Lu J, Kishore V, Gay G. Evaluation of genome-wide selection efficiency in maize nested association mapping populations., 2012, 124: 261–275.
[25] Lian L, Jacobson A, Zhong S Q. Genome wide prediction accuracy within 969 maize biparental populations., 2014, 54: 1514–1522.
[26] Technow F, Schrag T A, Schipprack W, Bauer E, Simianer H, Melchinger A E. Genome properties and prospects of genomic prediction of hybrid performance in a breeding program of maize., 2014, 197: 1343–1355.
[27] de Oliveira A A, Resende M F R Jr, Ferrão L F V, Amadeu R R, Guimarães L J M, Guimarães C T, Pastina M M, Margarido G R A. Genomic prediction applied to multiple traits and environments in second season maize hybrids., 2020, 125: 60–72.
[28] Wang X, Zhang Z L, Xu Y, Li P P, Xu C W. Using genomic data to improve the estimation of general combining ability based on sparse partial diallel cross designs in maize., 2020, 8: 819–829.
[29] Zhang A, Pérez-Rodríguez P, Vicente F S, Palacios-Rojas N, Dhliwayo T, Liu Y B, Cui Z H, Guan Y, Wang H, Zheng H J, Olsen M, Prasanna B M, Ruan Y Y, Crossa J, Zhang X C. Genomic prediction of the performance of hybrids and the combining abilities for line by tester trials in maize., 2021, 10: 109–116.
[30] 熊雪航, 段海洋, 李文龙, 李建新, 孙莉, 孙岩, 秦永田, 汤继华, 张雪海. 玉米穗长全基因组关联分析. 分子植物育种, 2022, https://kns.cnki.net/kcms/detail/46.1068.S.20220630.1359. 004.htmlXiong XH, Duan HY, Li WL, Li JX, Sun L, Sun Y, Qin YT, Tang JH, Zhang XH. Genome-wide association study of ear length in maize.,2022, https://kns.cnki.net/ kcms/detail/46.1068.S.20220630.1359.004.html (in Chinese with English abstract).
[31] 秦文萱, 鲍建喜, 王彦博, 马雅杰, 龙艳, 李金萍, 董振营, 万向元. 玉米叶夹角性状的全基因组关联分析与关键位点优异等位变异挖掘. 作物学报, 2022, 48: 2691–2705. Qin WX, Bao JX, Wang YB, Ma YJ, Long Y, Li JP, Dong ZY, Wang XY. Genome-wide association study of leaf angle traits and mining of elite alleles from the major loci in maize., 2022, 48: 2691–2705 (in Chinese with English abstract).
[32] 彭勃, 赵晓雷, 王奕, 袁文娅, 李春辉, 李永祥, 张登峰, 石云素, 宋燕春, 王天宇, 黎裕. 玉米叶向值的全基因组关联分析. 作物学报, 2020, 46: 819–831. Peng B, Zhao XL, Wang Y, Yuan YW, Li CH, Li YX, Zhang DF, Shi SY, Song CY, Wang TY, Li Y. Genome-wide association studies of leaf orientation value in maize., 2020, 46: 819–831 (in Chinese with English abstract).
[33] Pritchard J K, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data., 2000, 155: 945–959.
[34] Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure., 2007, 23: 1801–1806.
[35] Bradbury P J, Zhang Z W, Kroon D E, Casstevens T M, Ramdoss Y, Buckler E S. TASSEL: software for association mapping of complex traits in diverse samples., 2007, 23: 2633–2635.
[36] Wang J B, Zhang Z W. GAPIT Version 3: boosting power and accuracy for genomic association and prediction., 2021, 19: 629–640.
[37] Zhang Y W, Tamba C L, Wen Y J, Li P, Ren W L, Ni Y L, Gao J, Zhang Y M. mrMLM v4.0.2: an R platform for multi-locus genome-wide association studies., 2020, 18: 481–487.
[38] Cingolani P, Platts A, Wang L, Coon M, Nguyen T, Wang L, Land S J, Lu X, Ruden D M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3.(Austin), 2012, 6: 80–92.
[39] Luo Y, Zhang M L, Liu Y, Liu J, Li W Q, Chen G S, Peng Y, Jin M, Wei W, Jian L, Yan J, Fernie A R, Yan J B. Genetic variation incontributes to ear length and grain yield in maize., 2022, 234: 513–526.
[40] Ning Q, Jian Y N, Du Y, Li Y F, Shen X M, Jia H T, Zhao R, Zhan J M, Yang F, Jackson D, Liu L, Zhang Z W. An ethylene biosynthesis enzyme controls quantitative variation in maize ear length and kernel yield., 2021, 12: 5832–5842.
[41] Pérez P, de los Campos G. Genome-wide regression and prediction with the BGLR statistical package., 2014, 198: 483–495.
[42] Zhang Y M, Jia Z, Dunwell J M. The applications of new multi-locus GWAS methodologies in the genetic dissection of complex traits., 2019, 10: 100–105.
[43] An Y X, Chen L, Li Y X, Li C H, Shi Y S, Zhang D F, Li Y, Wang T Y. Genome-wide association studies and whole-genome prediction reveal the genetic architecture of KRN in maize., 2020, 20: 490–500.
[44] Zhou B, Zhou Z J, Ding J Q, Zhang X C, Mu C, Wu Y, Gao J Y, Song Y X, Wang S W, Ma J L, Li X T, Wang R X, Xia Z L, Chen J F, Wu J Y. Combining three mapping strategies to reveal quantitative trait loci and candidate genes for maize ear length., 2018, 11: 1–8.
[45] Li D D, Zhou Z Q, Lu X H, Jiang Y, Li G L, Li J H, Wang H Y, Chen S J, Li X H, Würschum T, Reif J C, Xu S Z, Li M S, Liu W X. Genetic dissection of hybrid performance and heterosis for yield-related traits in maize., 2021, 12: 774478–774496.
[46] Su C F, Wang W, Gong S L, Zuo J H, Li S J, Xu S Z. High density linkage map construction and mapping of yield trait QTLs in maize () using the genotyping-by-sequencing (GBS) technology., 2017, 8: 706–719.
[47] Chen L, An Y X, Li Y X, Li C H, Shi Y S, Song Y C, Zhang D F, Wang T Y, Li Y. Candidate loci for yield-related traits in maize revealed by a combination of MetaQTL analysis and regional association mapping., 2017, 8: 2190–2203.
[48] Zhou Z P, Li G L, Tan S Y, Li D D, Liu W X. A QTL atlas for grain yield and its component traits in maize ()., 2020, 139: 562–574.
[49] Zhao Y M, Su C F. Mapping quantitative trait loci for yield-related traits and predicting candidate genes for grain weight in maize., 2019, 9: 16112–16121.
[50] Lu X, Zhou Z Q, Yuan Z H, Zhang C S, Hao Z F, Wang Z H, Li M S, Zhang D G, Yong H J, Han J N, Li X H, Weng J F. Genetic dissection of the general combining ability of yield-related traits in maize., 2020, 11: 788–802.
[51] Galli M, Liu Q J, Moss BL, Malcomber S, Li W, Gaines C, Federici S, Roshkovan J, Meeley R, Nemhauser JL, Gallavotti A. Auxin signaling modules regulate maize inflorescence architecture., 2015, 112: 13372–13377.
[52] Koops P, Pelser S, Ignatz M, Klose C, Marrocco-Selden K, Kretsch T. EDL3 is an F-box protein involved in the regulation of abscisic acid signalling in., 2011, 62: 5547–5560.
[53] Zhang H H, Yin L L, Wang M Y, Yuan X H, Liu X L. Factors affecting the accuracy of genomic selection for agricultural economic traits in maize, cattle, and pig populations., 2019, 10: 189–198.
[54] Tehseen M M, Kehel Z, Sansaloni C P, Lopes M D S, Amri A, Kurtulus E, Nazari K. Comparison of genomic prediction methods for yellow, stem, and leaf rust resistance in wheat landraces from Afghanistan., 2021, 10: 558–572.
[55] Ma J, Cao Y Y. Genetic dissection of grain yield of maize and yield-related traits through association mapping and genomic prediction., 2021, 12: 690059–690069.
[56] Lozada D N, Mason R E, Sarinelli J M, Brown-Guedira G. Accuracy of genomic selection for grain yield and agronomic traits in soft red winter wheat., 2019, 20: 82.
[57] Odilbekov F, Armoniené R, Koc A, Svensson J, Chawade A. GWAS-assisted genomic prediction to predict resistance toBlotch in Nordic winter wheat at seedling stage., 2019, 10: 1224–1233.
[58] 马娟, 朱卫红, 丁俊强. 玉米重要农艺性状的基因组预测分析.玉米科学, 2022, 30(1): 48–52. Ma J, Zhu W H, Ding J Q. Genomic prediction analysis for maize important agronomic traits., 2022, 30(1): 48–52 (in Chinese with English abstract).
[59] Arruda M, Lipka A, Brown P, Krill A, Thurber C, Brown-Guedira G, Dong Y, Foresman B J, Kolb F L. Comparing genomic selection and marker-assisted selection for Fusarium head blight resistance in wheat ()., 2016, 36: 1–11.
[60] Bernardo R. Genomewide selection when major genes are known., 2014, 54: 68–75.
[61] Yuan Y, Cairns J E, Babu R, Gowda M, Makumbi D, Magorokosho C, Zhang A, Liu Y B, Wang N, Hao Z F, San V F, Olsen M S, Prasanna B M, Lu Y L, Zhang X C. Genome-wide association mapping and genomic prediction analyses reveal the genetic architecture of grain yield and flowering time under drought and heat stress conditions in maize., 2019, 9: 1919–1933.
[62] Liu Y B, Hu G H, Zhang A, Loladze A, Hu Y X, Wang H, Qu J T, Zhang X C, Olsen M, Vicente F S, Crossa J, Lin F, Prasanna B M. Genome-wide association study and genomic prediction of Fusarium ear rot resistance in tropical maize germplasm., 2021, 9: 325–341.
[63] Cericola F, Jahoor A, Orabi J, Andersen J R, Janss L L, Jensen J. Optimizing training population size and genotyping strategy for genomic prediction using association study results and pedigree information: a case of study in advanced wheat breeding lines., 2017, 12: e0169606.
Multi-locus genome-wide association study and prediction for general combining ability of maize ear length
MA Juan*, ZHU Wei-Hong, LIU Jing-Bao, YU Ting, HUANG Lu, and GUO Guo-Jun
Institute of Cereal Crops, Henan Academy of Agricultural Sciences, Zhengzhou 450002, Henan, China
Ear length is an important agronomic trait, which is closely related with yield. General combining ability (GCA) is an important index to evaluate excellent inbred lines. Therefore, the dissection of genetic basis of ear length GCA and formulation of corresponding breeding strategies is of great significance to improve maize yield. In this study, 537 F1hybrids as the experimental materials were obtained from 123 maize inbred lines and eight tester lines according to North Carolina II genetic mating design, and phenotyped under two environments. A total of 11,734 single nucleotide polymorphisms (SNPs) identified using the maize 5.5 K liquid breeding chip were used to conduct multi-locus genome-wide association study (MGWAS) and genomic prediction for ear length GCA in two environments and combined environment. A total of 11 SNPs significantly associated with ear length GCA were detected using seven MGWAS, and the variation of GCA effect explained by a single locus was 8.06%−28.23%. Five SNPs were co-located using different MGWAS. Locus 7_178103602 was repeatedly detected using mrMLM (multi-locus random-SNP-effect mixed linear model) in Zhoukou and combined environment, explaining 26.02%−28.23% of variation of ear length GCA, which was an environment-stable and major-effect SNP. 11 candidate genes were identified, among whichand EID1-like F-box protein 2 may be key genes for GCA of ear length. The accuracy of five random effect models for predicting ear length GCA ranged from 0.53 to 0.69 in the three environments, and there were minor differences among these models. In Xinxiang and Zhoukou environments, GBLUP (genomic best linear unbiased prediction) and RKHS (reproducing kernel Hilbert space) incorporating different significant loci as fixed effects could improve the accuracy of genomic estimated breeding value for GCA of ear length, with a percentage increase of 2.34%−14.98%. In the combined environment, except that the accuracy was slightly reduced using one significant locus derived from FarmCPU (fixed and random model circulating probability unification) or BLINK (Bayesian-information and linkage-disequilibrium iteratively nested keyway) as fixed effects, the addition of significant loci derived from the other two MGWAS methods could improve the genomic prediction ability, with a percentage increase of 2.80%−6.84%. Therefore, the incorporation of significant loci from MGWAS into the prediction models as fixed effects is helpful to improve the accuracy of the genomic estimated breeding value for ear length GCA, which could be used to effectively predict and select GCA of maize parental ear length.
ear length; general combining ability; multi-locus genome-wide association study; fixed effect model; genomic selection
10.3724/SP.J.1006.2023.23042
本研究由河南省科技攻关项目(222102110043)和河南省农业科学院优秀青年基金项目(2020YQ04)资助。
This study was supported by the Science and Technology Project of Henan Province (222102110043) and the Science-Technology Foundation for Outstanding Young Scientists of Henan Academy of Agricultural Sciences (2020YQ04).
马娟, E-mail: majuanjuan85@126.com
2022-05-18;
2022-11-25;
2022-12-05.
URL: https://kns.cnki.net/kcms/detail//11.1809.S.20221205.1216.001.html
This is an open access article under the CC BY-NC-ND license (http://creativecommons.org/licenses/by-nc-nd/4.0/).
猜你喜欢
麦类作物学报(2022年7期)2022-08-09
新世纪智能(数学备考)(2021年9期)2021-11-24
今日农业(2021年11期)2021-08-13
当代陕西(2019年15期)2019-09-02
学苑创造·A版(2018年11期)2018-02-01
读者(2017年5期)2017-02-15
江西农业学报(2016年5期)2016-06-21
江苏农业科学(2016年4期)2016-06-14
麦类作物学报(2016年4期)2016-05-27
遗传(2014年3期)2014-02-28
